The human pangenome is finally here and, in my opinion, it was the biggest story of the year
It's here! It's really here! The first draft of the human pangenome reference was released this year!
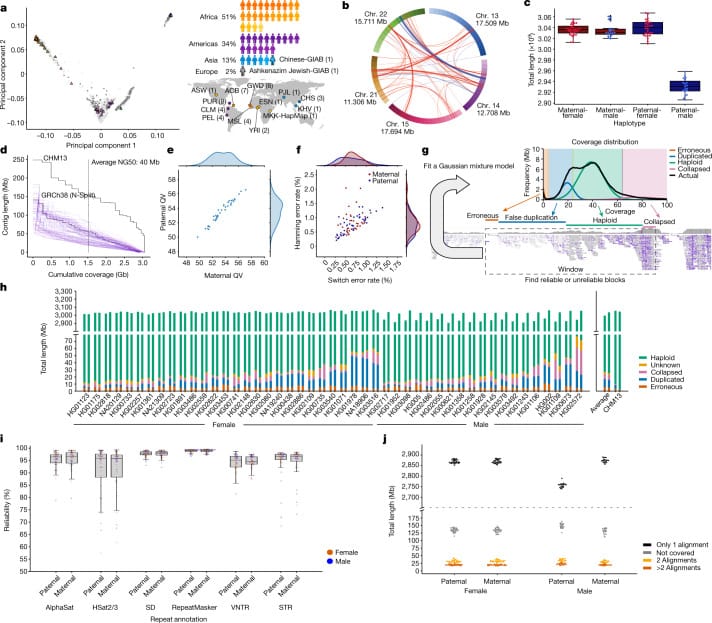
The Human Pangenome Reference Consortium (HPRC)'s first draft included 47 fully phased diploid assemblies of diverse individual genomes from 13 different ancestral backgrounds.

They assembled these genomes using predominantly long-read sequencing (PacBio and ONT) and genomic mapping (Hi-C and Bionano) with a dusting of Illumina Omni2.5 bead array genotyping and short-read for variant confirmation.
So why's it important that we now have 47 new, diverse, high quality genomes?
Mostly because despite sharing 99.9% of our genome with one another, there are still major differences in each of our genomes that we received from our specific ancestral lineage.
The first human genome was based mostly on a male of mixed race and did not account for all of the variation that we see across diverse populations which means it is severely lacking when it comes to helping us to determine which variants are causal of disease in different genetic backgrounds.
To fix this, the goal of the HPRC is to replace our dusty old linear reference genome with a graph genome that preserves ALL of the genetic diversity that we see across populations.
And if you don't believe that this is a big deal, let's dig in, because the benefits even with just 47 of the eventual 350 genomes are pretty impressive!
1) Variant Discovery - Showed improved performance, particularly in challenging regions and medically relevant genes - calling on average 64,000 more variants per 1kg sample and producing far fewer errors in both singletons and trios.
2) Genotyping Structural Variants - Detected significantly more SVs compared to short-read call sets, indicating that short-read SV discovery using linear reference genomes misses a significant proportion of SVs.
3) Analyzing Variable Number Tandem Repeats - VNTRs are regions in the genome that are very hard to sequence. The pangenome reduced mapping errors and enabled more accurate estimation of their length.
4) RNA-seq Mapping - Using the pangenome reduced false mapping rates, allelic bias, and increased mapped coverage on heterozygous variants, facilitating more accurate analyses of allele-specific expression.
5) ChIP-seq Analysis - Identified additional epigenetic marks that correlated well to pangenome specific structural variants with clear stratification of these marks between African and European populations!
Ultimately, the human pangenome will allow us to finally start to tease apart the complex, population-specific, genomic variants that account for all of the differences we see across populations.
While 47 genomes is a good start, I can't wait for the next 303 to be added in our quest to make genomic based healthcare more inclusive and equitable.