RhoFold+: A large language model for RNA structure prediction
Can LLMs predict RNA structures?
They certainly can try!
One of the biggest disappointments when AlphaFold 3 came out was that it wasn’t very good at predicting the structures of nucleic acids.
Despite it being billed as a structure prediction algorithm for all things biomolecules, the limitations of AlphaFold 3 and other structure prediction models has become increasingly apparent.
And as much as we want all of these things to work like a calculator, where the inputs produce a reliable (and correct) output, AI models aren’t quite there yet.
Although, it’s probably asking a lot for a model to ever be 100% accurate, because that’s the entire essence of a prediction model…sometimes they're wrong and you need to double check what they spit out (for forever).
But that doesn’t mean AI models are useless, or that they won’t get pretty close to perfection, it just means we need to do more iterations, feed them better data, and understand that a model isn’t validated until someone looks to see if the results it produces are correct!
One good way to do this is to benchmark the prediction models against known structures including structures that an algorithm has been trained on.
Because if it can’t get close to the right answer on a training set, there’s no way we’re ever going to be able to believe that what gets spit out for something completely new is worth following up.
This is especially true for things like RNA which can be extremely tricky to predict since nucleic acids can dynamically form many different structures.
But the structures formed by nucleic acids like RNA are important because they can affect how genes are expressed or even how RNA catalyzes reactions in our cells!
So, being able to accurately predict these structures is a big deal.
The authors of this week’s paper attempted to develop a model that could best AlphaFold (and others!) at predicting RNA structures and they named the tool they created RhoFold+.
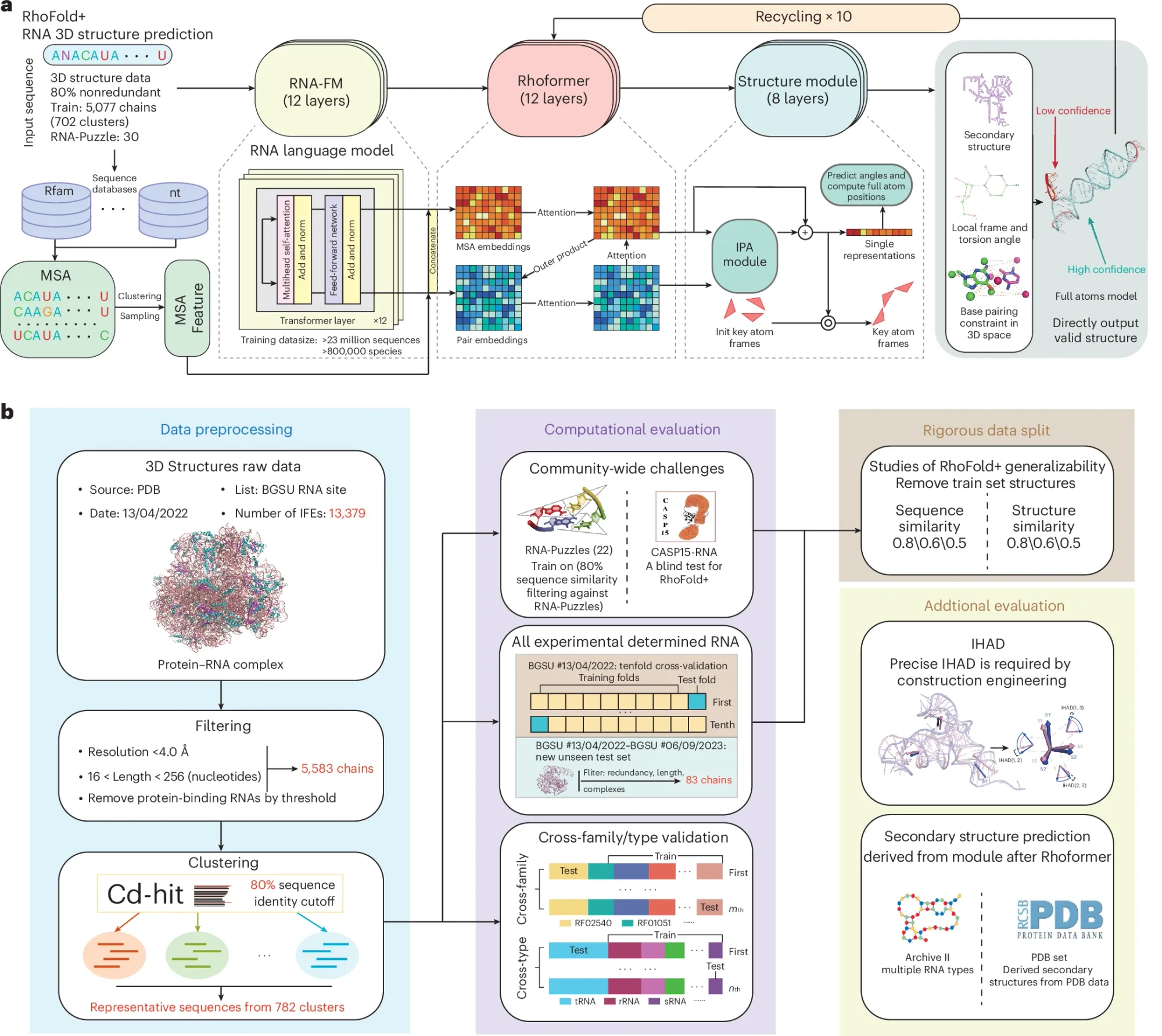
It’s described in the figure above:
a) It uses a large language model trained on RNA sequence and structure data that were then refined for ten cycles in their transformer network (Rhoformer) and finally that data went through the structural prediction module that assigns the locations and angles of the atoms in the RNA backbone.
b) Is a graphical abstract of the paper and provides an overview of how they selected the data that went into the model and how they validated its outputs.
Their benchmarking study showed that it performed significantly better than other RNA folding algorithms in the aggregate (but was not perfect), was less computationally intensive than its competitors, and its performance on experimentally determined structures could warrant its use on unseen structures.
But the authors were quick to highlight that the dynamic nature of RNAs make them hard to predict, so any work on unseen structures should be experimentally validated.