Omicly Weekly 7
January 14, 2024
Hey There!
Thanks for spending part of your Sunday with Omicly!
This week's headlines include:
1) The epigenome's role in aging gets some mechanistic answers
2) The Proteome: It's a bit more complex than any of us would like
3) Florence Bell and William Astbury's x-ray diffraction work on a 'pile of pennies'
Please enjoy!
Feeling old? It might be time to blame your epigenome.
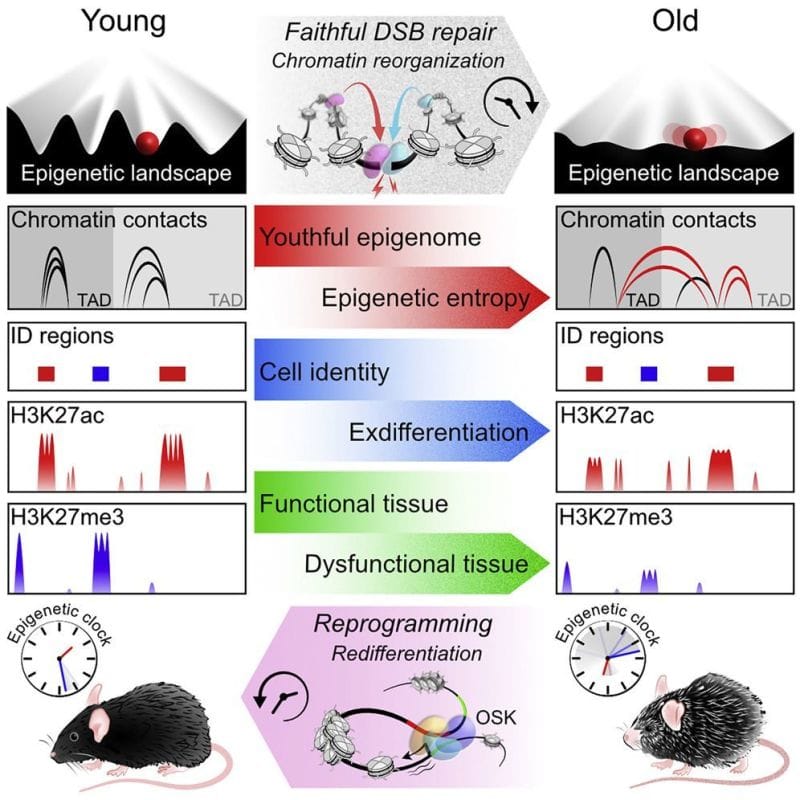
The link between DNA methylation, the epigenome and aging isn’t new.
We’ve known for some time that DNA methylation is important for controlling which genes get expressed.
DNA methylation, and epigenetics more broadly, are important for cellular differentiation and cell fate decisions during development.
Maintaining epigenetic marks is required for the proper function of tissues in multi-cellular organisms.
So, it stands to reason that disruption of those epigenetic marks as we age could have important consequences!
This is supported by evidence from studying aging in model organisms like yeast, fruit flies and naked mole rats.
These results seem to suggest that aging probably isn’t caused by the accumulation of DNA mutations over time.
Instead, changes to epigenetic factors, specifically those linked to regulating DNA methylation, have been shown to increase lifespan in many of these model systems.
Interestingly, these same factors have also been found to help to fix the genome when errors or breaks are found.
This makes sense, because we know that maintaining the epigenome is important for cellular function, so if there’s a break that needs to be repaired, the epigenetic marks in that repaired region also need to be re-established for proper function!
It’s estimated that up to 50 DNA breaks occur in every cell every single day, and over a cell’s lifespan, that’s a lot of repairs.
The author’s of today’s paper hypothesized that there was a link between these DNA repair functions and the loss of epigenetic control as we age.
They propose the ‘‘Information Theory of Aging’’ stating that, “aging in eukaryotes is due to the loss of transcriptional networks and epigenetic information over time, driven by a conserved mechanism that evolved to co-regulate responses to cellular damage...”
To test this hypothesis they developed a model system in mice where they could induce DNA strand breaks without causing mutations in any genes.
They quantified these effects molecularly by looking at changes in RNA expression, epigenetic regulatory marks and chromatin contacts.
Overall, the results show that these mice exhibit features of old age, and they link DNA repair to accelerated epigenetic aging.
Interestingly, the researchers showed in a follow-up experiment that activating Oct4, Sox2, and Klf4 (ES cell fans will be familiar with these already) in aged mice reversed the gene expression changes.
The main takeaway here being that excessive DNA damage repair and a loss of epigenetic information can cause aging but that this loss is potentially reversible.
Now we just need to find a safe and effective way to induce those epigenetic reset genes in every cell of our body.
###
Yang JH, et al. 2023. Loss of epigenetic information as a cause of mammalian aging. Cell. DOI: 10.1016/j.cell.2022.12.027
Proteoforms: This is where things start to get complicated.

You might be asking yourself, “WTF is a proteoform?”
The term ‘proteoform’ was first coined in 2013 by Lloyd Smith and Neil Kelleher.
It’s used to describe all of the different ‘forms’ of proteins that are created through the process of gene expression.
Our genomes contain information to code for approximately 20,000 genes.
That sounds like a big number, right?
Except, before we sequenced the human genome, we estimated that there should be about 100,000 genes to perform all of the complex functions that we see within our cells and tissues.
Oops.
So, how do we get 100,000 genes worth of effort out of 20,000 actual genes?
1) Alleles:
Our cells contain two copies of our genomes. One from our mom, one from our dad.
The differences between these two copies are referred to as alleles and they can produce slightly different proteins depending on the variants contained in each allele.
2) Alternative Splicing, Start, and Stop Sites:
Each gene is composed of exons and introns.
During the process of DNA transcription, the exons are connected together (spliced) to create messenger RNA and the introns are thrown away.
However, ‘alternative splicing,’ or the combination of different exons from the same gene, can create new isoforms.
This leads to the production of multiple different proteins from the same gene!
Over 90% of RNAs undergo alternative splicing and about 80% of genes have a minor isoform that represents 15% or more of expression from that gene!
Additional combinations of exons can be made by starting or stopping transcripts at different places which increases the combinatorial options even further!
3) Post-Translational Modifications (PTMs):
The process of converting that RNA message into protein is called Translation.
Once a protein chain is created it folds into its final structure and goes off to perform whatever function it needs to do in the cell.
God, I wish it was that simple...
Proteins are often post-translationally modified to control how they function, when they function, where they function or how well they function.
The most common of these modifications include:
Phosphorylation
Acetylation
Methylation
Glycosylation
Hydroxylation
Ubiquitination
Sumolyation
But this is by no means an exhaustive list and proteins can be modified with multiple different combinations of these (and other) PTMs.
‘Got it, but I'm still not clear on WTF a proteoform is?’
It is each unique version of a protein taking into consideration differences in a protein sequence, alternative splicing/Start/Stop and PTMs.
Theoretically, there are trillions of proteoforms for each gene (mostly due to all of the possible PTMs!).
But, practically (and in the proteins we’ve looked at closely), it’s in the tens to hundreds of proteoforms per gene.
This makes sense, because only the functional ones are going to be useful to a cell.
And that’s the overarching goal of proteomics: to figure out how all of these proteins work together to create biology and, ultimately, how they contribute to health and disease!
The first crystal structure of DNA was published in 1938. It was generated by Florence Bell, a scientist you need to know.
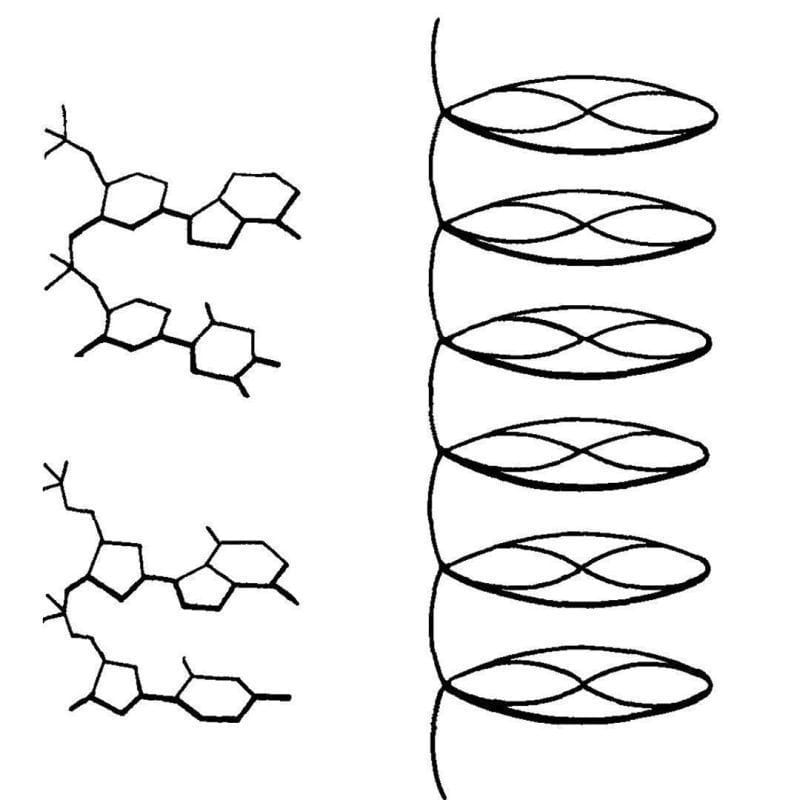
One thing that we lose in the coverage of major scientific discoveries is all of the work that those advances were built on.
A perfect example of this is Watson and Crick's solving of the structure of DNA in 1953.
This wasn't just a synthesis of data they obtained from Maurice Wilkins via Rosalind Franklin (and others), but it also wouldn't have been possible without the early work of William Astbury and Florence Bell.
This is that story.
In the 1930's, William Astbury spent his days analyzing biological fibers using a technique called x-ray crystallography.
He was particularly interested in coiled fibers like those found in wool (keratin) and other textiles and was the first to characterize the structural changes in proteins as they are stretched. He referred to these as the α-form and β-form, and laid the groundwork for Linus Pauling's discovery of the α-helix and β-strand in 1951!
Astbury quickly got a reputation for his expertise in x-ray crystallography, and was sent purified biological material to characterize from all over Europe.
In 1937, apparently elbows deep in things to characterize, he sent a letter to another renown crystallographer, Lawrence Bragg, asking if he knew of any rockstar crystallographers that were available to help with his studies.
Bragg recommended Florence Bell, a Cambridge grad who learned how to do crystallography from yet another x-ray luminary, JD Bernall. A short correspondence later and Bell left her job working for Bragg to start her graduate work under the tutelage of Astbury.
While her research was focused on lots of different biological fibers, Bell's most important work was with DNA.
She showed that the key to obtaining the most informative images was the use of high molecular weight DNA.
The best source for this was calf thymus, and the diffractions she obtained from these fibers indicated that DNA had an ordered structure.
This finding was published in 1938 and was also presented at Cold Spring Harbor later that year by Astbury. They described their structure of DNA as a 'pile of pennies' and a depiction of it can be seen above.
Their calculations also showed that the DNA nucleotides were 3.3 Å apart and they were stacked one on top of another.
While this might not seem like a historic achievement, it set the foundation for everything that would follow.
With a few tweaks and the discovery that DNA can exist in two forms depending on the humidity during crystallization, Rosalind Franklin and Raymond Gosling built on Bell's early work to generate photo 51 - the key diffraction of DNA used to solve the structure of DNA in 1953.
###
Astbury WT, Bell FO. 1938. Some recent developments in the x-ray study of proteins and related structures. CSHL Symp Quant Biol. DOI: 10.1101/SQB.1938.006.01.013