Omicly Weekly 5
December 31, 2023
Hey There!
It's the fifth and final Beta issue of Omicly!
I appreciate you signing up and sticking with me while I spent the last month figuring out how to create and send out a newsletter!
I think it's been pretty successful despite a couple early hiccups and thanks to everyone for all of the helpful suggestions.
Next Sunday will be the official launch so if you have any omic-ly inclined friends, please forward the newsletter to them so they can sign up!
Thanks for spending part of your Sunday reading my newsletter.
Please enjoy!
In this week's newsletter you will find:
1) Genomics is better with friends **cough** transcriptomics, proteomics, metabolomics, and epigenomics **cough**
2) Single-Cell and Spatial Transcriptomics and why they’ve gotten so much attention recently
3) The story of the polymerase chain reaction and how sometimes timing is everything
Spoiler Alert: The future of disease early detection isn't going to be genomics.

We already have some pretty good hints that this is true.
One of them is that many Multi-cancer early detection (MCED) screening tests are based on methylation.
This falls squarely in the realm of epigenomics.
But we also have three decades worth of high throughput genomics under our belts and we really don't have a ton to show for it.
We've certainly learned a lot in that time about the genome!
But one of the greatest lessons we've been taught is that genetics alone is terrible predictor of whether a healthy person will actually develop a disease.
Recent studies have shown that 8% of people carry a pathogenic mutation, but only about 7% of those people ever become symptomatic.
The caveat here being that some mutations are more likely to cause disease than others. These include mutations in BRCA1 and BRCA2 (Breast Cancer) or HBB (Thalassemia) in which 30-60% of patients with those develop disease.
So, what do we do with all of these 'damaging' pathogenic variants we find in healthy people?
The American College of Medical Genetics (ACMG) suggests extending the list of reportable findings to include pathogenic variants in 81 disease associated genes (inclusive of the 3 above).
These are all considered to be 'medically actionable' because steps can be taken to improve their clinical outcomes.
However, the struggle here, and with most genetic diseases, is knowing when, or if, a healthy person will ever become symptomatic.
Wouldn't it be great if we had a way to detect disease onset decades before there's a phenotype in a patient?
We're finally at a point where this might be possible and it's all thanks to new developments in proteomics (the study of proteins and how they interact within our cells)!
Interest in proteomics has increased drastically in the past few years as new technologies have made it possible to easily study thousands of proteins in a single sample.
The power of these methods was highlighted in papers throughout 2023.
But one in particular stood out where the plasma of 11,000 patients who had participated in a 25-year study were screened for ~5,000 proteins.
The researchers were able to use samples collected during that original study to identify a set of proteins that could predict the development of dementia decades before symptom onset.
That's pretty incredible!
While there's still a lot of work to be done, it's exciting to see how proteomics continues to increase our understanding of the biology of disease.
Because, as we've learned, the genome only represents potential, but the proteome could tell us when, or if, that potential is actually beginning to impact biology!
###
Walker KA et al. 2023. Proteomics analysis of plasma from middle-aged adults identifies protein markers of dementia risk in later life. Sci Transl Med. DOI: 10.1126/scitranslmed.adf5681
Single cell and spatial transcriptomics: What you need to know!

They're freaking cool!
The transcriptome is the study of all of the ribonucleic acid (RNA) produced within a cell.
It differs from the genome in a couple of important ways:
1) The transcriptome is made up of RNA messages, and the genome is made up of DNA
2) RNA is the template for making proteins, the genome is the template for making RNA
3) Each cell has the same genome but a different transcriptome
4) Each tissue (many cells working together) has a variety of transcriptomes that enable a function ie organ tissue
But for a very long time, our evaluation of transcriptomes were limited to surveying large populations of cells from ground up tissues or looking at whole blood.
We even have two consortiums dedicated to better understanding transcriptomes. One is the ENCODE Project or the ENCyclopedia Of Dna Elements and the other is the Genotype-Tissue Expression (GTEx) Project. Both projects have generated datasets that try to figure out how a single genome ends up creating the cellular diversity we see in all of our tissues by looking at regulatory elements in the DNA, protein associations with chromatin, epigenetic signatures, expression levels of RNA, and quantifying protein levels.
But, the initial datasets for ENCODE were based on sequencing homogenized tissues. This is a bit like trying to hang a picture with a sledgehammer when you don't have a tack hammer handy.
Thankfully, we can do more refined analyses now using single cell and spatial transcriptomics!
Single cell: This method was popularized academically as Drop-seq. It uses bead linked oil emulsions to capture transcripts from single cells for sequencing. Basically, a bead (or in the case of 10x Genomics' version, a hydrogel) covered in polyT capture sequences is combined in an oil droplet with a single cell. The oil droplet contains reagents that break open the cell and the polyT sequence hybridizes with the polyA sequence on the RNA transcripts. These sequences can be linked back to their original cells and grouped together because the capture sequence includes a cell barcode and a unique molecular identifier (UMI) sequence.
Spatial: The newest kid on the block which uses tissue samples as the input but instead of homogenizing these into single cells, they're treated more like a pathology slide. There are a couple of approaches here with some companies using capture array technology (10x), multiplex FISH (Vizgen), and UV microdissection (Nanostring) of selected regions of a tissue slide for capture and/or sequencing. They have the added benefit of being able to spatially profile protein locations using fluorescent antibodies; so not only can you use these systems to visualize where the RNAs are expressed in tissues but also the degree to which they create proteins. Which is super cool but also ends up producing some really beautiful images!
The method below has been cited more than 600,000 times and is one of the most important developments in the history of science.
The polymerase chain reaction (PCR) is used widely to amplify DNA sequences.
Kary Mullis is often given sole credit for developing PCR, which, according to him, he discovered while "riding DNA molecules" during an acid trip in the early 1980's.
But, today's story begins a decade earlier in the lab of Har Gobind Khorana.
Khorana won the Nobel Prize in 1968 for his work figuring out how RNA codes for protein.
The key to his success was that he and his team synthesized their own molecules of RNA, called oligonucleotides.
This allowed them to see what amino acids ended up in proteins after the translation of their sequences.
Khorana continued to focus on scaling this synthesis process to create a full gene sequence.
One of his post-docs, Kjell Kleppe, had an idea to use little pieces of complementary DNA to kickstart these synthesis reactions and copy a target sequence with DNA polymerases.
At the time, it was known that polymerases were involved in copying DNA during cell division and could use a 'primer' to start this reaction.
He presented his method and initial data in 1969 and published a paper where his method of in vitro "DNA repair replication" was described in 1971.
So, why does Mullis get all of the credit for the discovery of PCR?
Mostly because of timing.
In 1970, the Khorana lab was one of the few groups making oligonucleotides.
Kleppe's result was seen as interesting but technically infeasible to scale and the true power of DNA copying wasn't recognized until the early 1980's when cloning and other molecular manipulations were really taking off.
Mullis was employed at that time by Cetus Corporation and he stumbled on the idea of copying DNA using primers and polymerases all on his own.
He published an initial paper on PCR in 1985 using the E. coli ‘Klenow’ fragment (a truncated portion of DNA polymerase) which required manually cycling the reaction in a water bath and adding back enzyme every round because it wasn’t heat stable.
However, Mullis realized he could use the polymerase from a different bacteria, T. aquaticus, which lives in the boiling hot springs of Yellowstone Park.
This polymerase, which we now call Taq, was heat stable and didn't require replacement between cycles!
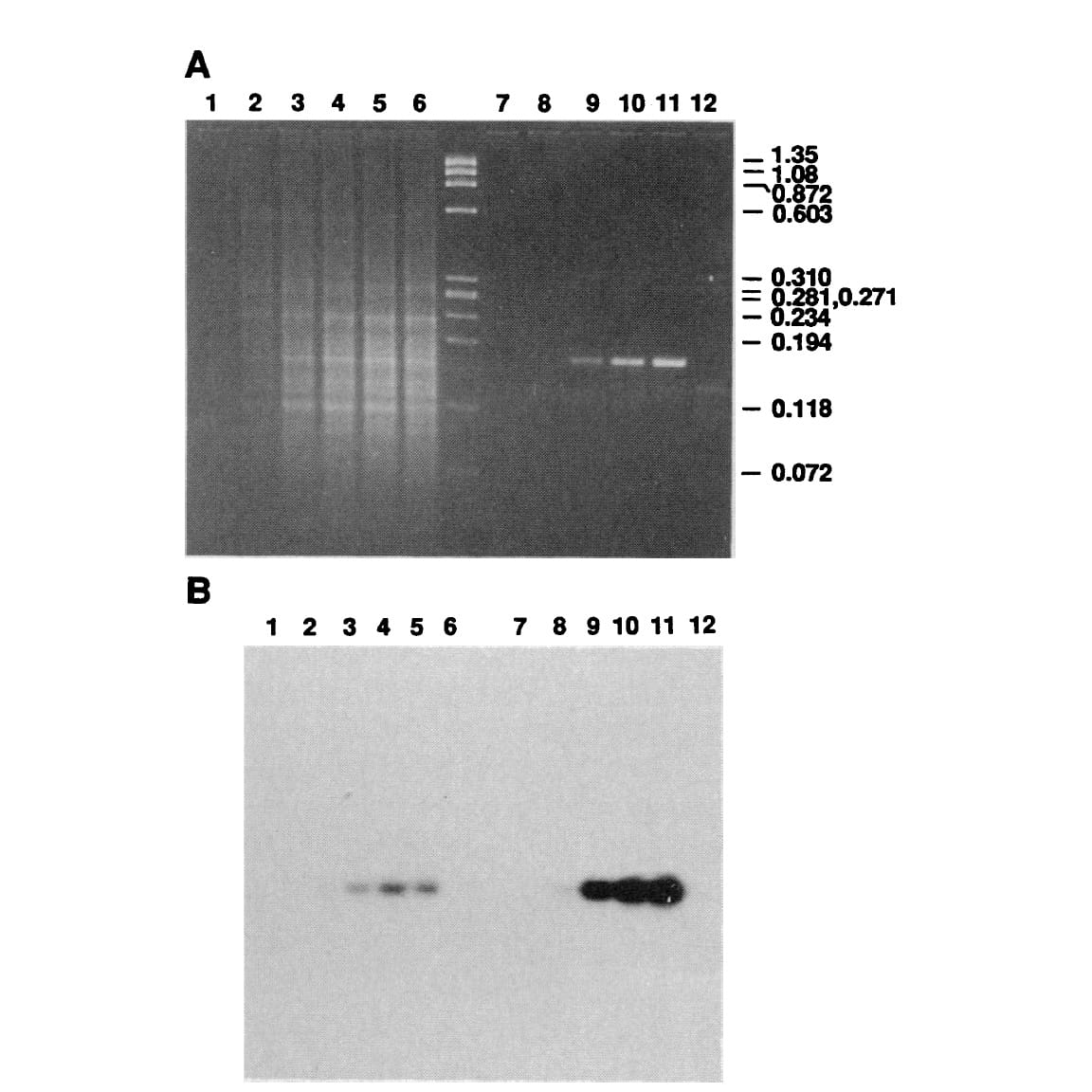
The figure above shows Mullis' comparison of Klenow (Lanes 2-5) to Taq (Lanes 8-11).
(A) is an agarose gel and (B) is a southern blot using a radioactive probe to detect the targeted DNA.
The benefits of using Taq are pretty obvious, but its heat stability was game changing because it made the process much cleaner and easy to automate!
Mullis received the Nobel Prize for this work in 1993.
###
Saiki RK, et al. 1988. Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science. DOI: 10.1126/science.2448875