Omic.ly Weekly 69
March 31, 2025
Hey There!
Thanks for spending part of your week with Omic.ly!
This Week's Headlines
1) Will nanopores enable single molecule protein sequencers?
2) Proteoforms make proteomics infinitely more complicated than genomics
3) Horseshoe crab blood to the rescue!
Here's what you missed in this week's Premium Edition:
HOT TAKE: 23andMe files for bankruptcy and is delisted from the NASDAQ signaling the collapse of array based consumer genetics
Brain storming about nanopore based protein sequencing generates new hypotheses to test
A new perspective in Nature Biotechnology recently highlighted the challenges of developing high throughput protein sequencers.
Sequencing proteins is difficult because the translation of mRNA to protein is the end of the line for our genetic code.
Francis Crick famously wrote in 'On Protein Synthesis' in 1958 that the Central Dogma was "that once ‘information’ has passed into protein it cannot get out again."
This has remained true, biologically, for the last 70+ years, but that's not stopping us from attempting to glean information from proteins via sequencing.
The concept of the central dogma is important when considering how we might go about sequencing proteins, because in the case of RNA and DNA, there exist enzymes we can co-opt to help with performing nucleic acid sequencing.
These include DNA, RNA and RNA dependent DNA polymerases (reverse transcriptases).
But no enzymes currently exist that can "read" a protein and spit out a sequence like theses polymerases do!
So we have to get creative if we want to sequence proteins!
But protein sequencing gets significantly more complicated than nucleic acid sequencing because proteins are built from 20 amino acids, not 4 bases, and each of those 20 amino acids can be modified to create alternative proteoforms!
So, for proteins, there's a lot more signal to deconvolute (and sometimes bonds to break) to get at the sequence.
There have been a number of proposals for how to do this and most of them come down to protein end degradation and recognition, "reverse translation," and direct sequencing with nanopores.
Protein end degradation based sequencing has been around for half a century or more, and you may have heard of "Edman degradation" which is a chemical method to chop amino acids off the ends of proteins.
There are at least two companies (Quantum-Si, Erisyon) working on low to medium throughput peptide sequencers that are based on degradation and recognition of terminal amino acids as peptides are cyclically degraded.
There are also companies (Encodia, Glyphic, AbrusBio) who have combined these degradation schemes with chemistry that "reverse translates" protein sequences into DNA sequences.
This is usually done with DNA oligo tagged antibodies that recognize specific amino acids and when these oligos come in close proximity to one another they're ligated together.
The protein sequence is then determined by sequencing all of these ligated oligos on a DNA sequencer.
But possibly the simplest, in theory, way to go about sequencing proteins is to look at them directly using nanopores!
We've figured out how to do this to sequence DNA and RNA, so the next logical step is to use nanopores to sequence proteins too.
But unlike DNA and RNA which have a strong negative charge, proteins do not have a single charge - regions of them can be positive, negative or neutral!
This makes DNA and RNA relatively easy to pull through a nanopore, but we have to get more creative if we want to use nanopores to sequence proteins.
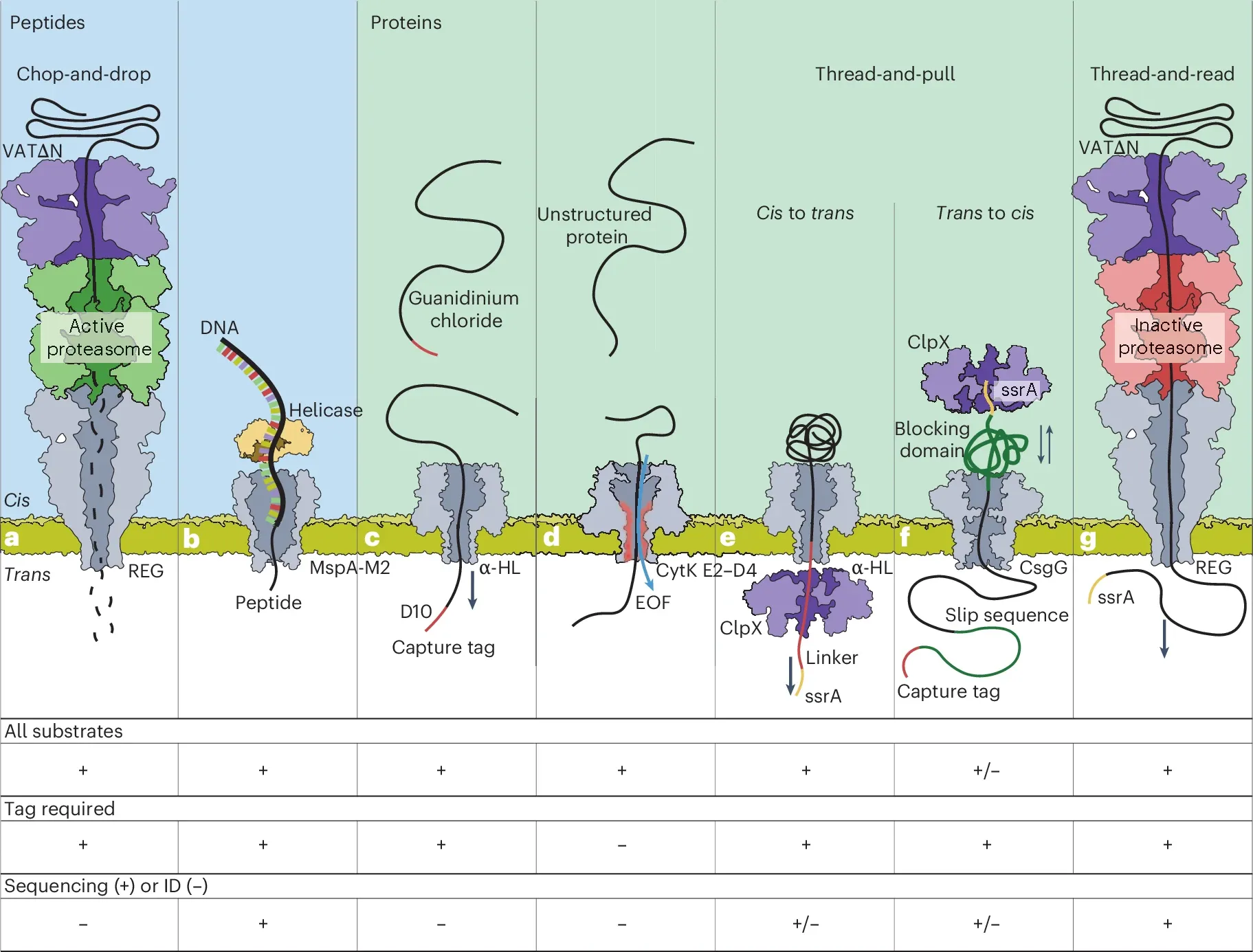
Some of these creative solutions can be seen in the figure above which reviews some proof-of-concept protein sequencers: a) an unfoldase (unfolds proteins) feeds an unfolded protein into a proteasome (chops up proteins) and peptides are detected as they pass through a pore b) a peptide is linked to a piece of DNA and threaded through a pore c) proteins are end tagged and pulled through a pore d) proteins are put in a special buffer and are pushed through a pore by electro osmotic flow (EOF) e,f) proteins are tagged and then either pulled (e) or pushed (d) through a pore by an unfoldase, and g) an unfoldase feeds a linearized peptide through a pore for sequencing.
All of these methods look interesting, but what I found most exciting about this paper (because I hadn't considered this before!) was how the authors proposed to go about evolving protein sequencing from "identification" to direct sequencing.
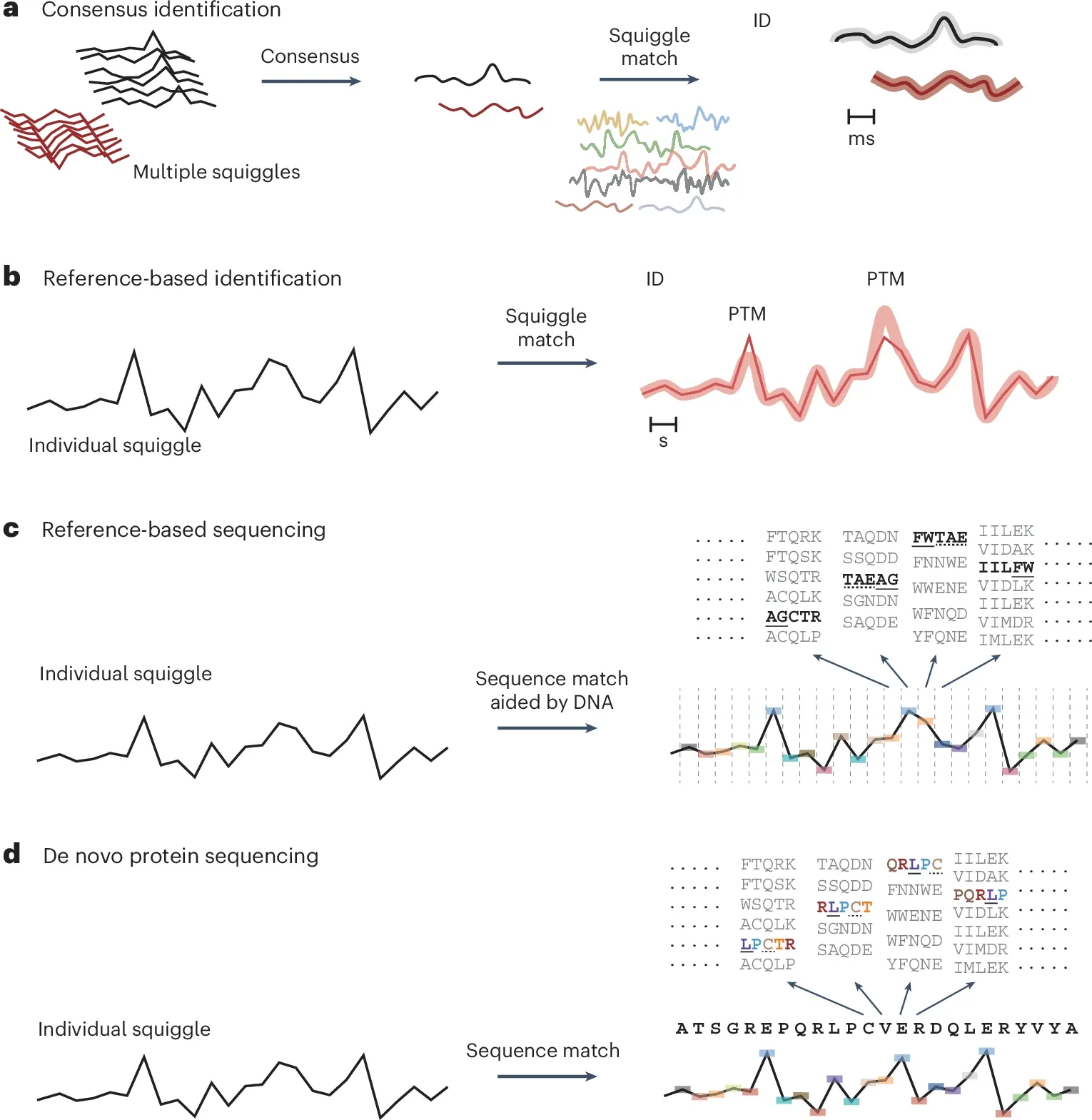
This is highlighted in the figure below where in a) they suggest building consensus squiggles (plots created as amino acids block the flow of current while transiting a pore!) for each protein to identify proteins and generate a reference library, b) use that reference library to identify proteins and make suggestions for where modifications might be residing based on changes in peak intensity, c) correlate squiggles to DNA to identify specific proteins, d) and as the technology further improves, identify amino acids and modifications directly.
A technological evolution like this means that nanopore based protein detection doesn't have to achieve perfection out of the box and can supplement the other imperfect methods we have now for protein detection like antibody/aptamer immunoarrays and mass spectrometry.
Whether nanopore based protein sequencers will ever be able to address the dynamic range problem (proteins come in varying abundance from one molecule to hundreds of thousands - detecting that wide of a range is hard!) remains to be seen, but 10 years ago some thought that nanopore DNA sequencing would be impossible.
I'm hoping for similar innovations in nanopore protein sequencing that will silence the naysayers once again.
###
Lu C, et al. 2025. Toward single-molecule protein sequencing using nanopores. Nature Biotech. DOI: 10.1038/s41587-025-02587-y
Proteoforms: This is where things start to get complicated.

You might be asking yourself, “WTF is a proteoform?”
The term ‘proteoform’ was first coined in 2013 by Lloyd Smith and Neil Kelleher.
It’s used to describe all of the different ‘forms’ of proteins that are created through the process of gene expression.
Our genomes contain information to code for approximately 20,000 genes.
That sounds like a big number, right?
Except, before we sequenced the human genome, we estimated that there should be about 100,000 genes to perform all of the complex functions that we see within our cells and tissues.
Oops.
So, how do we get 100,000 genes worth of effort out of 20,000 actual genes?
1) Alleles:
Our cells contain two copies of our genomes. One from our mom, one from our dad.
The differences between these two copies are referred to as alleles and they can produce slightly different proteins depending on the variants contained in each allele.
2) Alternative Splicing:
Each gene is composed of exons and introns.
During the process of DNA transcription, the exons are connected together (spliced) to create messenger RNA and the introns are thrown away.
However, ‘alternative splicing,’ or the combination of different exons from the same gene, can create new isoforms.
This leads to the production of multiple different proteins from the same gene!
Over 90% of RNAs undergo alternative splicing and about 80% of genes have a minor isoform that represents 15% or more of expression from that gene!
3) Post-Translational Modifications (PTMs):
The process of converting that RNA message into protein is called Translation.
Once a protein chain is created it folds into its final structure and goes off to perform whatever function it needs to do in the cell.
God, I wish it was that simple...
Proteins are often post-translationally modified to control how they function, when they function, where they function or how well they function.
The most common of these modifications include:
Phosphorylation
Acetylation
Methylation
Glycosylation
Hydroxylation
Ubiquitination
Sumolyation
But this is by no means an exhaustive list and proteins can be modified with multiple different combinations of these (and other) PTMs.
‘Got it, but I'm still not clear on WTF a proteoform is?’
It is each unique version of a protein taking into consideration differences in a protein sequence, alternative splicing and PTMs.
Theoretically, there are trillions of proteoforms for each gene (mostly due to all of the possible PTMs!).
But, practically (and in the proteins we’ve looked at closely), it’s in the tens to hundreds of proteoforms per gene.
This makes sense, because only the functional ones are going to be useful to a cell.
And that’s the overarching goal of proteomics: to figure out how all of these proteins work together to create biology and, ultimately, how they contribute to health and disease!
Horseshoe crab blood is our first line of defense against endotoxin contamination of surgical tools and medical devices. Seriously.
We figured out early on that sticking unsterilized medical equipment into people during surgeries usually ends badly.
And badly here means that a lot of people died from bacterial infections.
"Isn't that why we use an autoclave to sterilize those things?"
Yes, but that's only half of the solution.
Because gram negative bacteria also produce endotoxins like Lipopolysaccharide (LPS) which make up their cell wall.
Autoclave sterilization kills bacteria but has no effect on LPS which is a potent stimulator of the immune system and can lead to septic shock.
This can be caused by the Shwartzman Phenomenon which was first discovered by Gregory Shwartzman in 1926.
He found that injecting rabbits with two doses of gram negative bacteria (have an endotoxin laden cell wall) but not gram positive bacteria (no endotoxin) led to severe, hemorrhagic necrosis at the injection site - or, the immune system reacted, clotted the area, and caused the cells in that location to die.
You can see why endotoxin contamination of medical devices and other things that make contact with blood might be a bad thing.
So, for a long time, device manufacturers kept large colonies of rabbits and used them as endotoxin detectors.
At the end of manufacturing, devices would be washed with a saline solution and that would then be injected into a rabbit. If the rabbit got a fever, it meant the device was contaminated.
However, the rabbits got a reprieve in 1956 when Frederick Bang made a crazy discovery while studying the horseshoe crab Limulus polyphemus.
One of his crabs got sick, became lethargic and died. Upon closer inspection, he noticed that the crab's blood turned into a thick gel.
Doing what any curious scientist would, he cultured the blood, saw gram negative bacteria (endotoxins!), and injected the cultures into a bunch of other crabs.
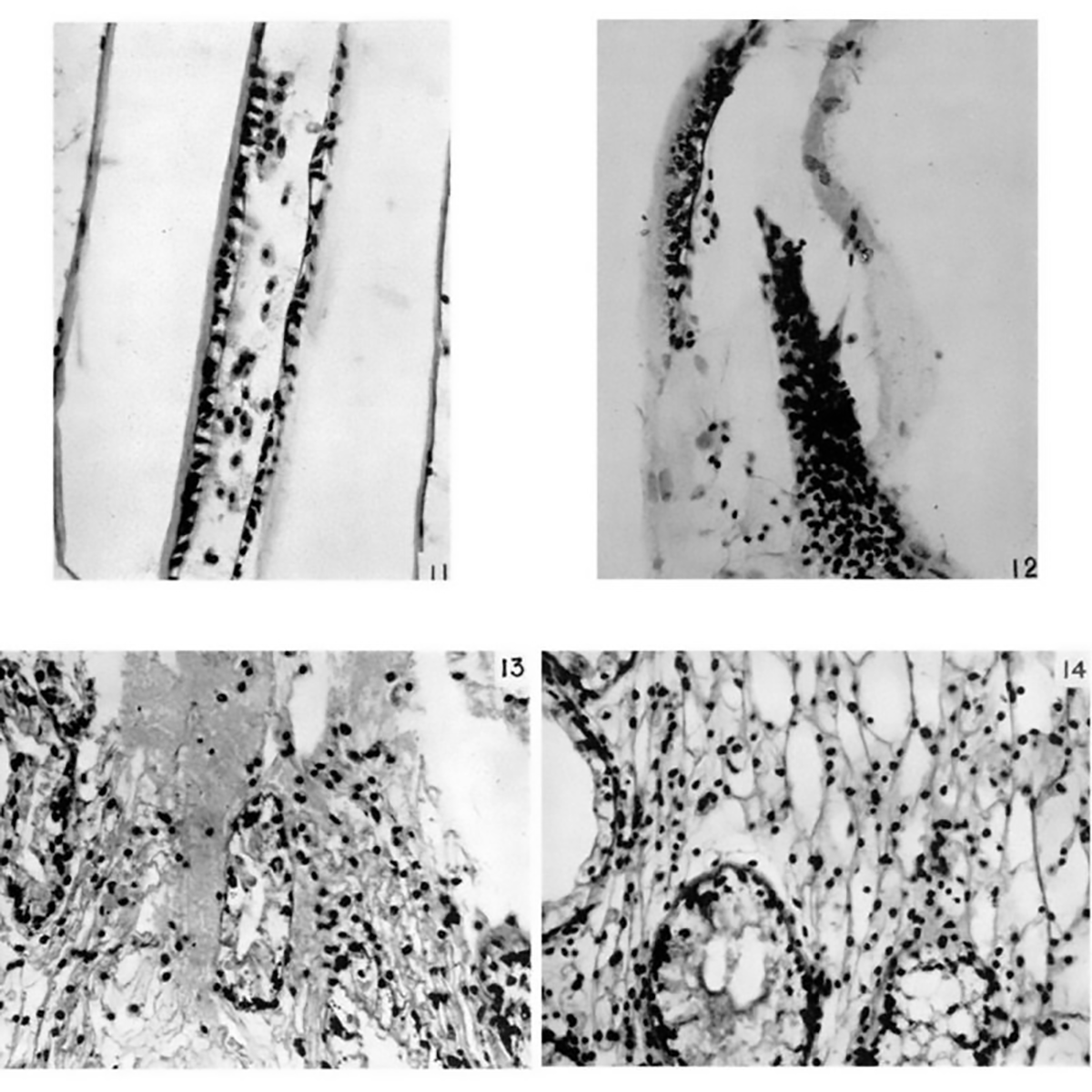
Today's figure is a compilation of the 'before and after' of those injections. Figures 11 & 13 are 'before,' and 12 & 14 are the clotted 'after.'
Bang, thinking this looked a lot like the Shwartzman Phenomenon on steroids, hired Jack Levin, a hematologist (blood nerd) to study this effect further.
What Bang and Levin discovered was that horseshoe crab blood cells, called amoebocysts, are highly sensitized to endotoxins and secrete a massive amount of clotting factor in an attempt to stop an infection!
It was quickly realized that a test based on horseshoe crab blood would be WAY cheaper than maintaining rabbit colonies and in the 1970's the FDA approved the Limulus Amoebocyte Lysate Assay for this use.
More recently, regulatory changes were proposed in 2023, enabling the use of a synthetic version of this test, and should reduce the bleeding burden of our crab friends.
###
Bang FB. 1956. A bacterial disease of Limulus polyphemus. Bull Johns Hopkins Hosp. PMID: 13316302
Were you forwarded this newsletter?
LOVE IT.
If you liked what you read, consider signing up for your own subscription here: