Omic.ly Weekly 67
March 17, 2025
Hey There!
Thanks for spending part of your week with Omic.ly!
This Week's Headlines
1) The creation of the (adorable) Woolly Mice-mmoth!
2) Just because it's in your genes, doesn't mean it's your fate
3) The formulation of the histone code forever changed our view of the genome
Here's what you missed in this week's Premium Edition:
HOT TAKE: Turnabout is fair play as Illumina gets caught up in a trade war kerfuffle
Or if you already have a premium sub:
Colossal Biosciences makes a mini-mammoth mouse using multiplex gene editing
Colossal Biosciences bills itself as a de-extinction company with the goal of resurrecting long-lost animal species like the woolly mammoth.
Although, that seems to maybe not be an accurate picture of what they're actually doing, which is mutating genes in extant (not extinct) animals to make them LOOK LIKE an extinct animal.
Those are two very different things.
But resurrecting extinct animals, especially ones that weren't well preserved, or that are thousands of years old, is impossible.
This is because the half-life of DNA is about 500 years, so anything older than 500 years is going to have a genome that's a jumbled mess unless it was preserved under pristine conditions.
But even our Woolly Mammoth friends who have been trapped in the Siberian Tundra for eons weren't preserved well enough for us to extract some nuclei and Dolly up a copy 🐑.
So, the next best thing is to make an animal that looks like the thing that went extinct.
How does one go about making a Woolly Mammoth look-a-like?
Well, you start with an elephant, and then give it long, flowy locks!
At least, that's the plan, but elephants are tough customers when it comes to making genetically modified versions because they have a 22-month gestation period.
Waiting 2 years to see if what you did worked isn't exactly a rapid turn around.
So, Colossal have instead done a bunch of their early editing work in mice!
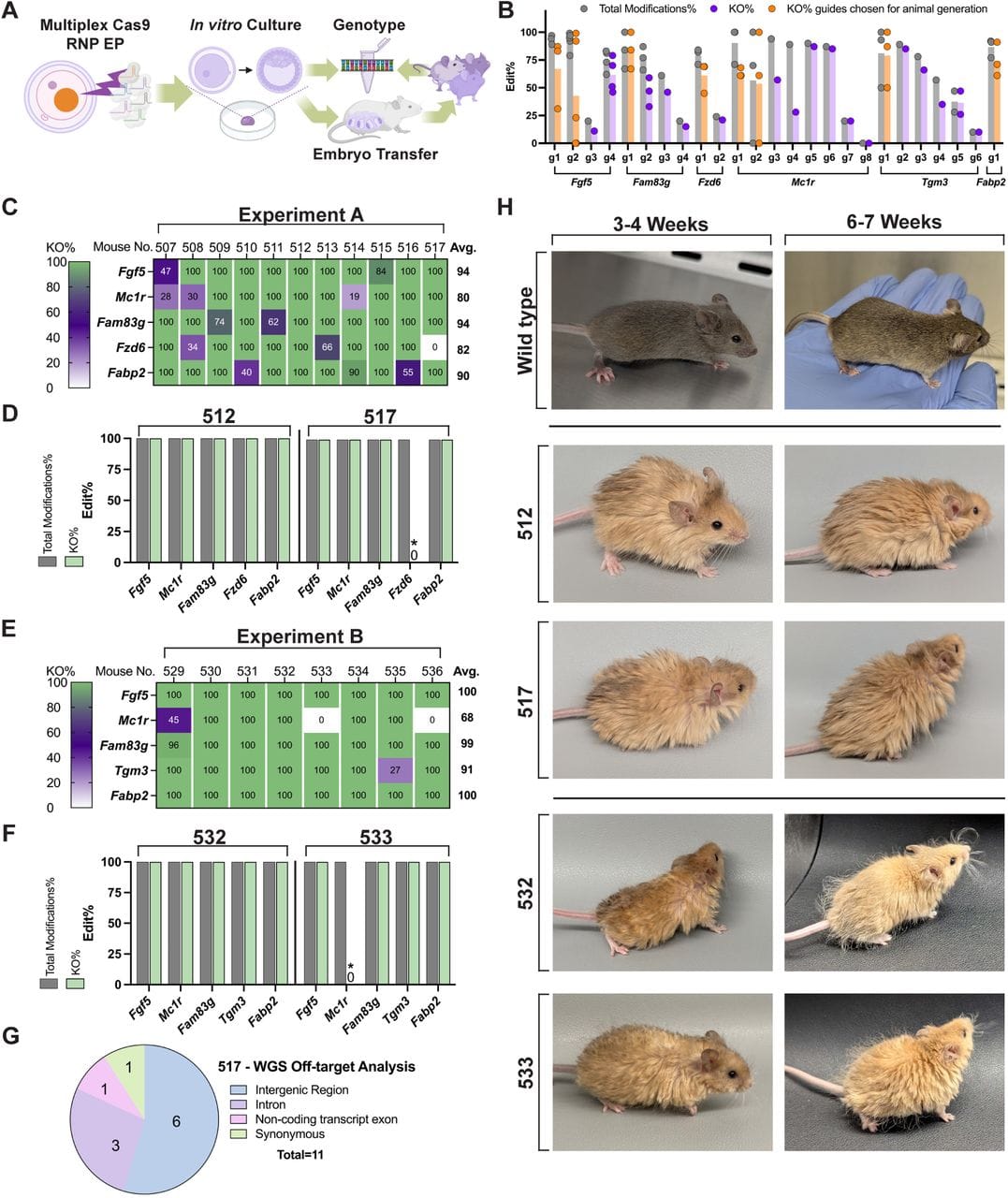
What they've come up with can be seen in the figure above where they multiplex edited a mouse to give it a Woolly appearance.
They selected a number of genes already known to produce abnormal hair morphology in mice and combined them together with a couple of other genes observed to be mutated in mammoth genomes.
A) is an overview of how they did the CRISPR editing, B) shows a graph of how they screened for the best guide RNAs (CRISPR/Cas uses these to make their targeted cuts) - the orange ones are the ones they used for multiplex editing, C,D,E,F) the researchers performed two editing experiments and the efficiency of the editing in each embryo line can be seen in the tables and charts, G) shows there were few off target effects and none that damaged other genes, and H) is the pièce de résistance which displays the cute little fur balls that were created!
Colossal went on to show that they can also use cytosine base editing to get similar results.
This paper is important because genome engineering has come a long way since the early days of CRISPR, and this work highlights that it can now be highly efficient, rapidly producing animals with a desired phenotype.
And, while all the chatter about "de-extinction" is fun, that's not what Colossal is doing here.
What they've actually done is develop a multiplex editing platform to create chimeric animals and cells for use in agriculture or the pharmaceutical industry.
This is still interesting, but we shouldn't pull the wool over people's eyes about bringing extinct animals back into existence.
###
Chen R, et al. 2025. Multiplex-edited mice recapitulate woolly mammoth hair phenotypes. Biorxiv (Preprint). DOI: 10.1101/2025.03.03.641227
One thing we constantly battle in genomics is, “What does it all mean?”

As with everything, it's complicated.
We have 5 million variants in each of our genomes with about 100,000 of those falling in the coding regions of genes.
Of those, a couple hundred are damaging but the majority are what we refer to as variants of unknown significance (VUS).
The process of determining whether those variants cause disease (are pathogenic) or benign (don't cause disease) isn’t incredibly sophisticated.
It’s generally done by looking at families with a history of disease, finding what variants are common in those affected and then deciding with some degree of certainty based on the function of a gene whether or not a variant fits well with the symptoms.
One of the most used databases for assessing whether a variant might cause a disease is ClinVar, and underpinning the sophistication of this process, ClinVar ranks the certainty of a classification with gold stars.
Yes, your kindergarten teacher was onto something after all.
So, it’s probably fair to say we’re still pretty early in the game of figuring out all of this stuff, but too often in genomics we try to find signal in the noise.
Because if there’s a variant, it has to DO something!
And unfortunately, this means we stumble into the pernicious problem of genetic determinism or this idea that the code written in your genome is the only thing that determines whether you will develop a disease.
We realized a while ago that genetics really is only a component here and other genetic, environmental and epigenetic factors all contribute to whether a disease develops (this is called penetrance) and similarly, these factors can also determine the severity of disease in an individual (this is sometimes referred to as expressivity).
However, historically, these measurements were done in families who were affected by a genetic disease so the estimations of penetrance have always been assumed to be over estimates.
Today, we can do better because we now have huge population level datasets.
Recent studies have found that about 8% of people carry a pathogenic mutation, but only 7% of those were symptomatic in the EHR.
However, the penetrance of individual mutations varies greatly, for example, on average, 38% of BRCA mutation carriers develop breast cancer.
These results highlight the importance of approaching disease risk associations carefully, especially when reporting results in healthy people.
The genome only ever represents what 'could be,' but observing changes in our other 'omes,' which are the functional product of the genome, can help us better predict when genetic variations might actually manifest as disease.
Because genetics isn't deterministic, and for us to truly get a handle on disease and disease progression, we need to marry what we see in the genome with what we can measure in the other 'omes.'
Histones were once thought to just be dumb structural proteins that held chromatin together; now we know that the ‘histone code’ is a major regulator of gene expression.
The first studies on gene expression were done in bacteria.
Here, gene expression is very straightforward:
Proteins bind to DNA, turn it into RNA and then that RNA gets read to create other proteins.
Things get much trickier in complex organisms.
Their larger genomes are packaged into a super-structure called chromatin.
And, in 1974, Roger Kornberg proposed that chromatin was made up of a DNA-protein complex that he named the ‘nucleosome.’
DNA is wrapped around histone proteins to create this highly ordered structure but this also presents an interesting problem to solve.
If DNA is all coiled up, how does the gene expression machinery get in there to keep cells alive?
Fortunately, David Allis suspected that histones were critical players in gene regulation.
He remembered earlier work from Vincent Allfrey showing that histones could be methylated or acetylated and he was determined to figure out if these modifications performed a function.
His chosen model for this was the single-celled protozoan Tetrahymena.
This might seem an odd choice, but Tetrahymena are full of acetylated histones and have high levels of gene expression which made them perfect for the task!
Allis and his team figured an enzyme was responsible for all of this acetylation and isolated the first histone acetyltransferase (HAT) from them in 1996.
They found that this enzyme, p55, was similar to Gcn5p, a protein known to regulate gene expression in yeast.
This discovery, along with additional work from Michael Grunstein, connected HATs to gene activation.
But adding an activation mark to histones only solves half of the gene expression problem.
How do you turn genes off?
Fortunately, the first histone deacetylase (removes acetyl groups) was discovered soon after by Stuart Schreiber and this explained how acetylation levels were maintained in cells.
Following these two discoveries, additional histone modifying enzymes were found that added phosphorylation and methylation to the mix.
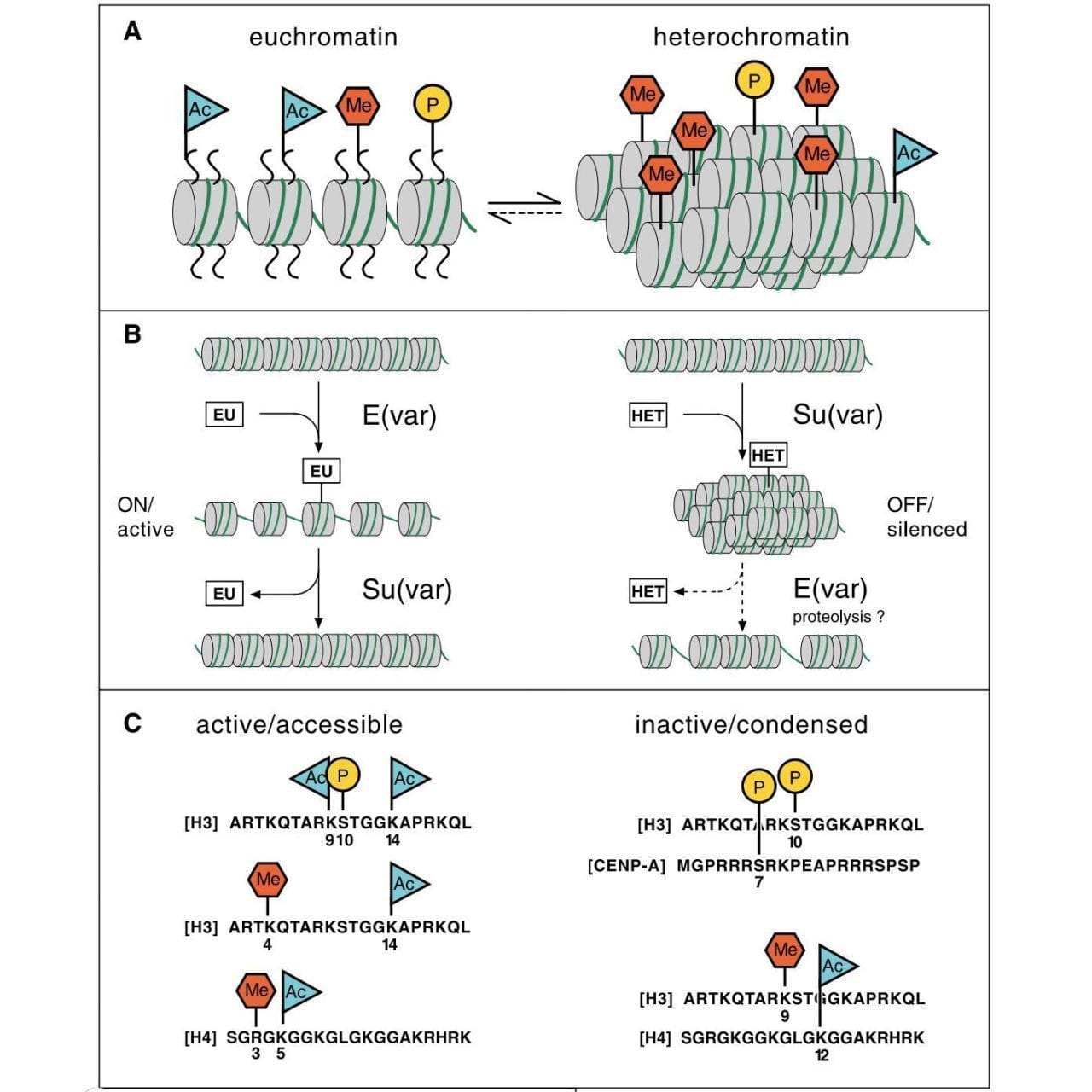
The figure above pulls all of this together into what is known as the histone code, or, how histone modifications are added, read, and erased to control what regions of DNA are accessible for gene expression.
A) displays the histone marks associated with open (euchromatin) and closed (heterochromatin), B) shows a model of how suppressors and enhancers of variegation work to open and close chromatin, and C) shows which amino acids on each histone are marked in open and closed chromatin.
But, this review article is important because it is one of the first to link histone modifications to epigenetics and this idea fundamentally changed how we viewed gene regulation at the chromatin level.
###
Jenuwein T, Allis CD. 2001. Translating the histone code. Science. DOI: 10.1126/science.1063127
Were you forwarded this newsletter?
LOVE IT.
If you liked what you read, consider signing up for your own subscription here: