Omic.ly Weekly 65
March 3, 2025
Hey There!
Thanks for spending part of your week with Omic.ly!
This Week's Headlines
1) New genes and pathways have been discovered in bipolar disorder
2) How genomic mapping finally helped to "complete" the human genome
3) The structure of tRNA won a Nobel, the person who figured it out, didn't
Here's what you missed in this week's Premium Edition:
HOT TAKE: Ultima gets cheaper, Roche collaborators cheer real-world use cases of SBX, Illumina deflects to a "robust pipeline" and spatial biology at AGBT 2025
Or if you already have a premium sub:
60-80% of people with bipolar disorder (BD) inherited it from a family member, but the genetic causes of BD are largely unknown.
BD, like schizophrenia and other psychiatric disorders, is highly heritable but we've struggled to pinpoint exactly what genes are involved in causing it.
BD comes in two forms: Both are characterized by having bouts with depression but BDI includes mania (abnormally elevated mood or behaviors) and BDII includes hypomania (slightly less elevated, but still elevated, mood/energy).
But our inability to identify genetic causes in BD is pretty frustrating considering the number of people afflicted with the disorder and the fact that genetic tests, like high throughput whole genome sequencing, are now available at a reasonable cost.
Historically, the field of psychology has relied heavily on performing genome wide association studies (GWAS) in these individuals because this technology is cost effective, and after spending years trying to find a single gene cause for BD (and not finding many), it's been assumed that the disorder is polygenic.
Describing a disorder as polygenic just means that we think more than one gene is contributing to the development of the disorder.
This makes it significantly harder to understand the cause and how to treat it.
Psychiatry, and GWAS in general, also struggles with the fact that most studies on these disorders are performed in white Europeans so the contributions of genes in more diverse populations has been ignored.
This potentially has made us miss important contributors and pathways involved in the development of BD.
However, a recent meta analysis of 158,036 individuals with BD and 2,796,499 controls across European, East Asian, African American and Latino ancestries has found 298 new genetic loci linked to BD.
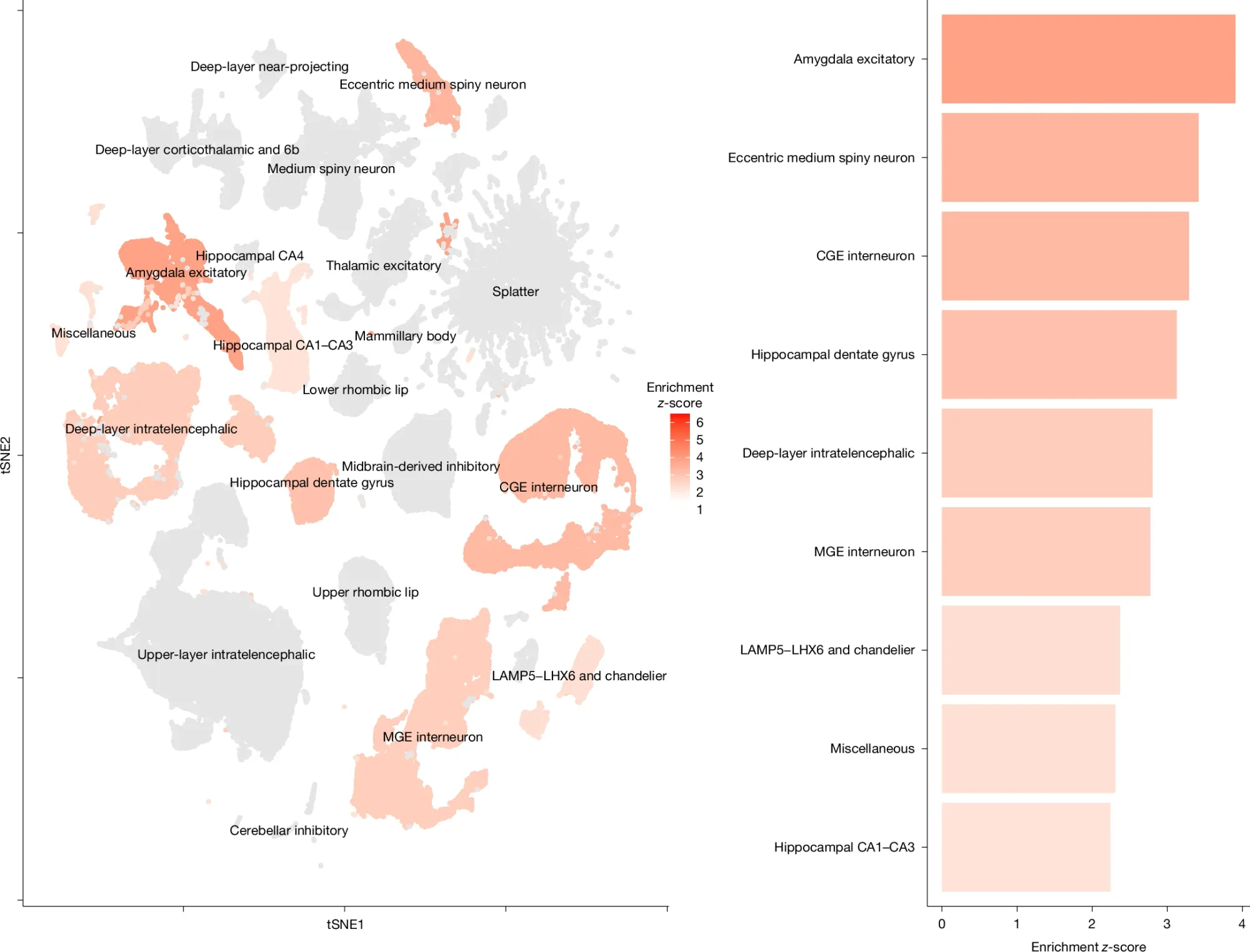
In the figure above, a gene set enrichment analysis was performed and correlated with a recent single-cell RNAseq dataset to determine which neuronal subtypes are most likely to be affected in BD (gray, not associated, orange to red, associated).
The clustering of the data indicate that synaptic neurons, prefrontal cortex and hippocampal interneurons, and hippocampal pyramidal neurons could be important for the development and progression of BD.
In addition to the neuronal cell type association data, they found 36 new genes associated with BD and a drug targeting analysis indicated that at least 16 of them could be good candidates for small molecule therapy.
This study identifies new genetic and neurobiological mechanisms underlying BD which could be used in the development of new treatments or therapies.
However, because this was a meta analysis of SNP based GWAS datasets, there's likely other (rarer or ancestry specific) associations that would be found using more comprehensive genomic methods.
###
O’Connell KS, et al. 2025. Genomics yields biological and phenotypic insights into bipolar disorder. Nature. DOI: 10.1038/s41586-024-08468-9
Did you know the human genome wasn't actually completed until 2022? No, seriously!

The Human Genome Project declared in 2003 that the human genome was completed, but it had a Dwayne Johnson sized hole in it since it actually only covered ~90% of the genome.
Back when the human genome was sequenced, old school Sanger sequencing was used which had a maximum effective read length of ~800-1500bp.
But even 800bp wasn’t long enough to orient the highly repetitive stretches of DNA found at the tips and the centers of chromosomes.
These regions were not completely resolved until early 2022 using long-read sequencing and genomic mapping (optical mapping and Hi-C).
But this fact raises an important point about the current state of genomics.
The organization of our chromosomes and the locations of genes within them controls how the cells in our bodies use and access that genetic information.
While short-read sequencing is very good at determining the order of those bases, it's not very good at finding the larger structural arrangements of the genome that span thousands (kilo) or millions (mega) of bases.
This means they miss important variants like inversions, translocations, or those that fall in repetitive elements.
So, how can we better understand the longer range organization of the genome?
Historically, this has required us to actually, physically, look at chromosomes with a microscope via karyotyping.
While this works, this process is technically challenging and requires us to catch cells in Metaphase. This is the phase of the cell cycle where chromosomes compact into their classical X structures - like 99% of the time, chromosomes actually look like a giant ball of yarn, which isn’t very useful for viewing purposes
But genomic mapping allows us to get useful information out of those giant balls of yarn with much better resolution than classical imaging techniques.
Optical Genomic Mapping (OGM): Optical maps are generated by restriction enzyme labeling and then threading ultra high molecular weight DNA through what looks like a miniature PLINKO machine. This stretches out and linearizes the DNA. The fluorescent labels are then imaged and aligned with one another to create genomic maps.
Hi-C: A genome wide sequencing technique that captures the conformation of chromosomes. Basically, when chromatin (DNA and structural proteins) is in that ball of yarn, it comes into contact with other parts of itself. These interactions can be 'fixed' with formalin and then detected by sequencing the fragments that were stuck together. Interaction heat maps turn into genomic maps because sequences closer to each other will be 'hotter' since they 'touch' more often.
But, as with every new development, when you look at the genome in a way that you haven't looked at it before, you find things.
And that means we're probably going to hear a lot more about structural variants in the coming years.
The next great mystery to solve after the discovery of the DNA double helix was to figure out how the nucleotide sequence coded for proteins.
This was a challenging concept in the 1950's because prior to the publication of the Hershey-Chase experiment in 1952, there was a dispute over what the genetic material actually was.
Linus Pauling and other prominent scientists believed that the genetic material was protein.
They reasoned that since there were 20 amino acids, protein was obviously the genetic material because DNA only had 4 nucleotides and lacked the complexity to store information!
However, Alfred Hershey and Martha Chase showed with the help of a blender full of bacteria (and phages) that DNA, and not protein, was unequivocally the genetic material.
This presented a significant problem.
Watson and Crick get credit for solving the structure of DNA in 1953 (using data from Rosalind Franklin), but Crick's later theoretical work postulating how 4 nucleotides could code for all 20 of the amino acids was just as important.
In a lecture in 1957, he laid out 3 hypotheses (and the central dogma!):
1) RNA is the intermediary between DNA and protein
2) Certain RNAs serve as amino acid adaptors
3) These 'adaptor RNAs' use a triplet code to translate the genetic information into protein
Concurrently and without knowledge of Crick's hypotheses, Paul Zamecnik and Mahlon Hoagland discovered that small, soluble, RNAs could be charged with amino acids through an enzymatic process in 1958.
This independently confirmed Crick's adaptor hypothesis.
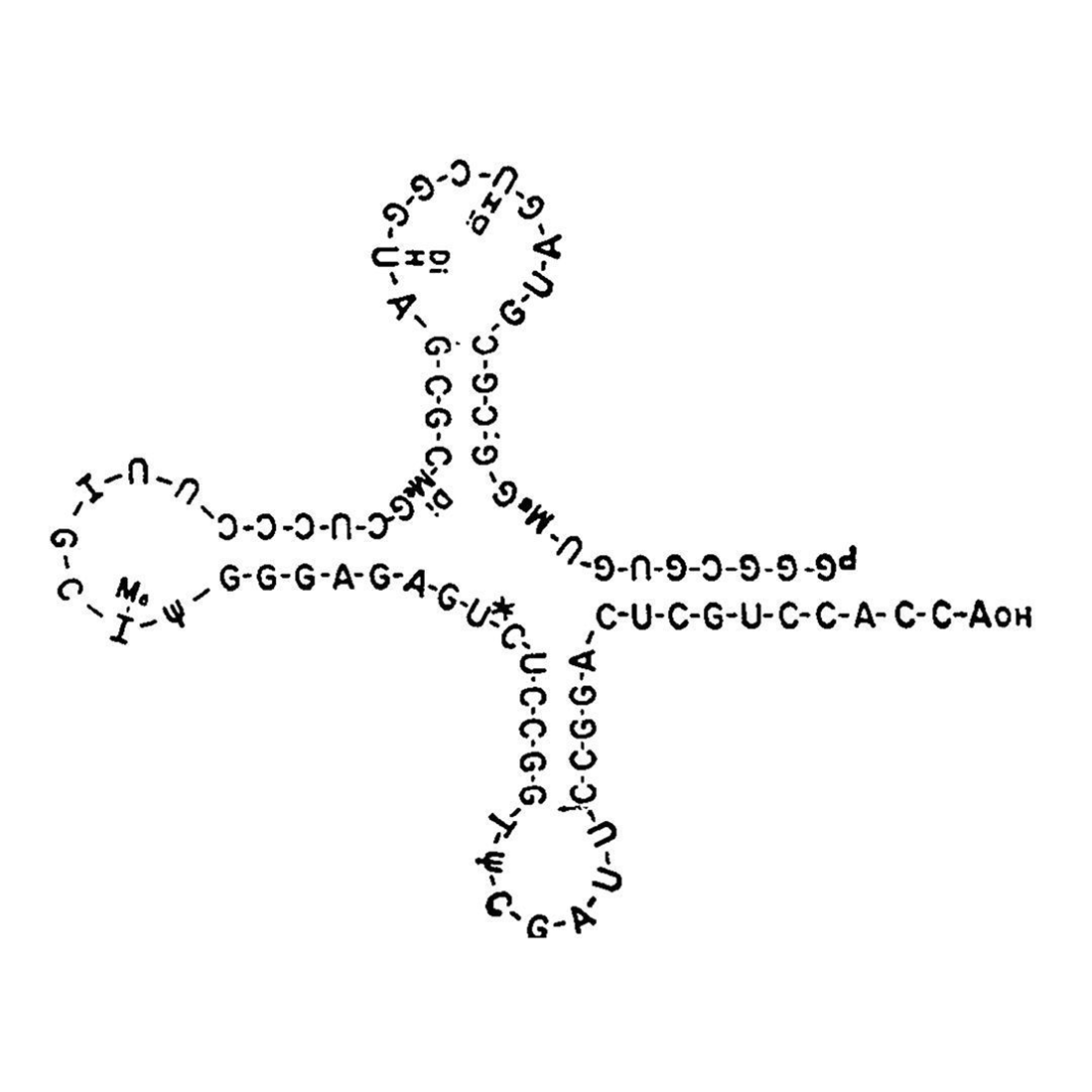
However, it was Robert Holley and his team at Cornell that determined the structure of this amino acid charged RNA which we now refer to as transfer RNA or tRNA.
Holley and team spent 9 years methodically sequencing tRNA using pancreatic and Taka-Diastase ribonucleases to chop up the RNAs into small fragments and then piece them back together.
They published the complete structure of all 77 nucleotides of tRNA-ala in 1965.
Ultimately, its the structure of tRNA that explains how it functions in the translation of RNA.
Holley shared the Nobel Prize with Khorana and Nirenberg, who deciphered the triplet code, in 1968.
The figure above is the cloverleaf structure of tRNA depicted in that 1965 publication.
It was proposed by a member of Holley's team, Elizabeth Beach Keller.
She was acknowledged for 'helpful suggestions.'
While her name does not appear as an author nor is she credited in textbooks that contain this now famous structure, her 1997 obituary tells the story of how she modeled tRNA using pipe cleaners, paper and velcro.
She sent the final structure to Holley in a Christmas card.
Her friend, Joseph Calvo, remembered her work during that time, "Her contributions were always behind the scene, looking until she found the shape that something wanted to take.''
###
Holley RW et al. 1965. Structure of a Ribonucleic Acid. Science. DOI:10.1126/science.147.3664.1462
Were you forwarded this newsletter?
LOVE IT.
If you liked what you read, consider signing up for your own subscription here: