Omic.ly Weekly 63
February 17, 2025
Hey There!
Thanks for spending part of your week with Omic.ly!
This Week's Headlines
1) MRD Testing: It's just a phase
2) Karyotypes? FISH? Arrays? What is this, the 20th century???
3) Telomerase and how linear genomes attempt to stay long
4) Weekly Reading List
Here's what you missed in this week's Premium Edition:
HOT TAKE: Dante Genomics plays buzzword bingo, adding Omics and AI to their new moniker
Or if you already have a premium sub:
All of the variants are getting in on MRD testing, even the phased ones!
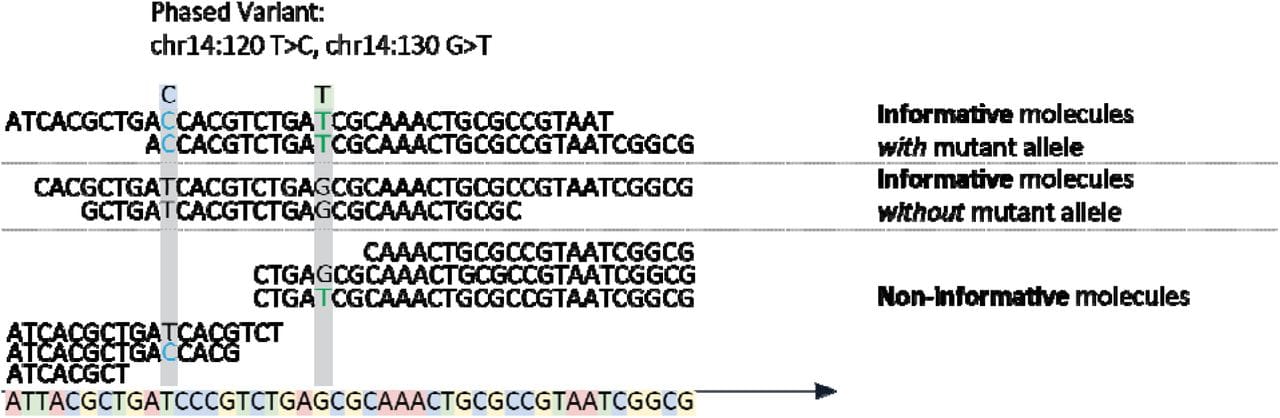
Minimal Residual Disease (MRD), also known as Molecular (when looking at nucleic acid) and Measurable (because, why not make this confusing?) Residual Disease describes the amount of cancer left in the body after treatment.
Measuring MRD over time allows us to know when a cancer might be coming back.
It also can help us make decisions about what treatments to use, or tell us if an adjuvant therapy (treatment, like chemo, given after a tumor is removed) is working.
But determining residual disease in patients has historically been pretty challenging.
And, up until the invention of massively parallel high throughput sequencing, knowing when or if a cancer might recur was almost impossible.
That’s changed now that the price of sequencing has come down drastically in the last two decades.
It’s now cheap enough to use routinely to help us find early clues about cancer’s recurrence.
We can do this because normal cells and cancers dump some of their DNA into the bloodstream as they grow.
These DNA fragments can be sampled in a blood draw and the “cell free” or “circulating tumor” DNA (cfDNA or ctDNA) can be sequenced to determine if the fragments are regular old normal DNA or DNA that came from a cancer.
This is usually done by looking for single nucleotide variants (SNVs) and comparing those variants to a person’s healthy normal tissue.
Fragments found to have differences could indicate that a cancer is present.
Or they could just be random errors that get introduced during the process of manipulating a sample.
These errors, unfortunately, can sometimes end up producing a false-positive result.
So, we inevitably end up sequencing a bunch of DNA to try find all of the needles (ctDNA) in the haystack of DNA that’s circulating in our blood (cfDNA) to be really really sure we haven’t made any mistakes.
But wouldn’t it be nice if we had a better way to determine which fragments of DNA came from tumors, and which were just the product of weird errors made by the enzymes that we use to process these samples?
We’ve invented a couple of interesting solutions here that involve clever barcoding schemes, but these can add significant cost to the process.
A group of researchers recently wondered if they could do this more simply using phased variants to both reduce the false positive rate and track tumor DNA within the bloodstream.
Phased variants are just variants that occur on the same DNA fragment.
And because cancers mutate frequently, they often have more phased variants in their genomes than are found in healthy tissue.
With this in mind, the researchers behind today’s paper developed a method called Phased Variant Enrichment and Detection Sequencing (PhasED-Seq) and validated its use as an MRD test in large B-cell lymphoma (DLBCL).
Here, the test starts by sequencing tumor and healthy DNA to develop a tumor specific phased variant (PV) list.
That list is then used for subsequent monitoring of plasma samples to see if those same PVs reappear in the bloodstream!
The figure above shows the basics behind the method which involves the capture of DNA fragments from regions known to be frequently mutated in DLBCL followed by short-read high throughput sequencing to detect PVs.
Informative molecules are those that span two phased variants, uninformative ones are those that span the region but only contain one or none of the variants.
In their analytical validation, they show that their test using PVs had a very low false positive rate (0.24%), a limit of detection of 0.7 parts in 1,000,000 and a precision of more than 96%.
That’s pretty good!
And with so many of these high performing MRD tests coming to market, it will be exciting to see how these new tests will help to extend the lives of cancer patients.
###
Boehm N, et al. 2025. Analytical Validation of a Circulating Tumor DNA Assay using PhasED-Seq Technology for Detecting Residual Disease in B-Cell Malignancies. BioArxiv Preprint. DOI: 10.1101/2024.08.09.24311742
Genomic mapping: bringing cytogenetics into the 21st century

One of the biggest subspecialties in clinical genetics is cytogenetics.
It is the field within pathology that deals with cellular anatomy as it relates to chromosomes.
For the longest time, humanity’s best guess at figuring out genetic disease was either to look at the physical attributes of a person (phenotype) or look at the chromosomal super-structures within cells using a microscope.
Cytogenetics today is mostly focused on pre-natal/post-natal clinical testing in fetuses and infants.
These are some of the first tests that are performed when there's a suspected genetic disease.
You might be thinking, "We have sequencing now, so what's the point of looking at chromosomes?"
Because structural variants like inversions, translocations and deletions/duplications are a big deal, but doing anything using sequencing in cytogenetics is a very recent development and this is underscored by the fact that up until about 2005, archaic methods like metaphase karyotyping were still the gold standard cytogenetic technique.
But, in the 21st century, we can do better, faster, cheaper with far higher resolution!
For comparison, the traditional methods for looking at chromosomes are:
Karyotyping: Either stain based banding or more recent molecular banding based techniques with a resolution of about 5 megabases (mb).
FISH: Fluorescent in-situ hybridization which is where fluorescently labeled probes are used to detect specific chromosomal abnormalities or paint whole chromosomes.
aCGH: Array comparative genomic hybridization uses probe array technology to detect genomic anomalies with 50-100kb resolution.
"But what about non-invasive prenatal testing, isn't that sequencing???"
Yes, but it’s not diagnostic, and the resolution of NIPT is on it’s best day 5mb. It also cannot reliably detect inversions or translocations - because, short-reads.
"Ok, then what about whole genome sequencing?"
Same problem as above but with slightly better resolution. If you're doing WGS anyway, short-read based cytogenetic predictions can RULE IN a diagnosis but they can't be used to rule out one because short-reads miss things.
Enter high resolution genomic mapping.
The leader in this space is Bionano Genomics and they use two enzymes to fluorescently tag DNA.
The DNA is then stretched out, linearized, and imaged in megabase sized chunks.
The little florescent tags can then be used to reconstruct, or map, a genome with a resolution of 3-10kb, picking up everything that's missed by competing methods for a couple hundred dollars.
Nabsys now does something similar with a pore based detection scheme.
Or, if you have a penchant for doing sequencing, you can spend a bit more to use Hi-C based techniques (Arima, Phase Genomics, etc) to make similar reconstructions.
But the biggest barrier here for broad clinical use will be convincing payers to reimburse for them!
What if I told you that the ends of our chromosomes are a paradox and we didn't fully understand how human DNA was copied until 1989?
Meselson and Stahl showed in 1958 that replication of the DNA double helix, the process that copies DNA, is semi-conservative.
This means that the daughter double helix contains one newly synthesized strand and one old strand that serves as the template for copying.
During the process of replication, the parent DNA helix is unwound at an 'origin of replication' and the copying process begins.
Unlike humans, and other organisms that have a linear genome, bacteria don't have to figure out how to copy the ends of their genomes!
Bacteria have a circular genome so the process finishes where it began and ends with the creation of two circles.
But, if a genome is linear, there's a big problem when the machinery hits the end of a chromosome.
You see, replication only occurs in a single direction, 5'->3'. There's a 'leading strand,' which is made contiguously as the DNA is copied, and a 'lagging strand,' which is made by creating short fragments that are connected together using an enzyme called DNA ligase.
The problem is that once the leading strand gets to the end of a chromosome, the machinery that simultaneously creates the lagging strand doesn't have enough DNA at the end to sit on to fill in that last little fragment!
The lagging strand should always end up being just a little bit shorter than the leading strand, and so, a genome should get shorter every time it's copied!
Except it doesn't.
And that's due to a repetitive sequence at the end of our chromosomes called the telomere!
But we didn't really know what the telomere did until 1975 when Elizabeth Blackburn discovered that a highly repetitive nucleotide sequence (C-C-C-C-A-A) made up the telomeres of tetrahymena (a freshwater microorganism).
Blackburn had an inkling that this repetitive sequence was important and for the next 14 years she, along with help from Jack Szostak and Carole Greider, figured out how the ends of chromosomes are copied and preserved.
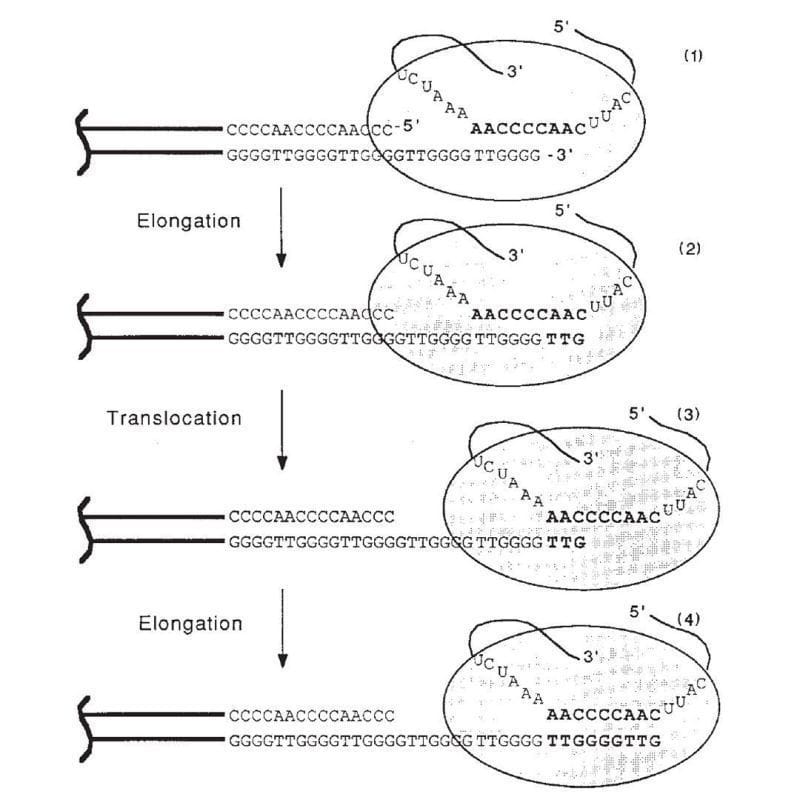
The figure above is the culmination of those years of experiments.
1) Shows the end of a chromosome and the telomerase enzyme (oval) with its RNA template bound to the TTGGGG of the telomere. Telomerase is a reverse transcriptase - these use RNA as a template to create DNA.
2) Elongation occurs, adding a TTG to the telomere.
3) Telomerase shifts down the strand (translocates) 6 bases to the new TTG.
4) Elongation occurs again.
This process of elongation and translocation is repeated multiple times, adding back sequence and preventing the shortening of the genome during replication!
Blackburn, Greider and Szostak were awarded the Nobel Prize for this discovery in 2009.
###
Greider CW, Blackburn EH. 1989. A telomeric sequence in the RNA of Tetrahymena telomerase required for telomere repeat synthesis. Nature. DOI:10.1038/337331a0
Were you forwarded this newsletter?
LOVE IT.
If you liked what you read, consider signing up for your own subscription here: