Omic.ly Weekly 62
February 10, 2025
Hey There!
Thanks for spending part of your week with Omic.ly!
This Week's Headlines
1) BONUS HOT TAKE: Indirects, what are they good for?
2) Rare diseases get a second opinion and a new diagnoses
3) FFPE Sucks.
4) The discovery that X-inactivation is caused by a long non-coding RNA
Here's what you missed in this week's Premium Edition:
HOT TAKE: The US Patent Trial and Appeal Board earmarks 10x Genomics' single-cell patents for a one-way trip to the dumpster
Or if you already have a premium sub:
BONUS HOT TAKE: Do we seriously need to have a conversation about why indirects are important? Really?!
You may have heard that this week the Trump NIH has decided to immediately cap the indirect rate for all federal research grants at 15%.
But this isn't our first time down this path!
They tried and failed to lower it to 10% in 2017.
Their reasoning this time is that private foundations provide lower indirects and that reducing the indirect rate will "curb leftist agendas."
I hate to break it to my conservative friends, but science is not a leftist agenda.
That's not to downplay the important conversation we need to have about indirect rates which help to cover things like facilities, electricity, water, administrators, and funding the purchase of all of the equipment that end up in core facilities at universities.
Maybe you think there's administrative bloat, or that institutes spend too much money.
You might also be upset that some institutions get 30% indirects while other schools get close to 80%. (The average is 27%)
Why is there such a discrepancy?
Well, title II part 200 of the code of federal regulations (we'll get back to why this is important) says that indirects are calculated by taking the cost of doing science at the university (facilities, administrators, etc) and dividing that by how much money they spend directly doing research (the direct costs).
But universities don't just get this money, they have to submit documentation to support their calculations and then negotiate the rate with the federal government.
So, research institutions set their budgets based on what they've negotiated with the federal government which is determined by the laws of our country.
And they use those budgets to plan things like building facilities, or buying equipment that amortize/depreciate over decades.
Just capping the indirect rate at 15% without warning is a seriously damaging change that imperils not only the budgets of our top research institutions, but also all of the ongoing and future scientific work that they are doing.
Can a president and his associates just say that the laws of the US don't apply anymore?
Probably not, and I'm certain that this will go to the courts and be blocked like many of the other illegal actions that have been undertaken in the last few weeks.
But I want to try to instill in all of you the importance of federal research grants that are provided to our public and private universities.
The most innovative things we are doing right now are happening at these research universities.
It is the discoveries that these institutions make that turn into the products that we all use today.
And it is our research universities that do the important, high risk research that serve as the backbone for all of the advances that we have seen in healthcare, pharma, aerospace, energy and every other high technology industry in existence.
Investment in biomedical research alone has over a 2x ROI and generates billions of dollars in economic activity.
The US is unquestionably the most scientifically advanced nation on this planet and it's because we invest so heavily in these discovery efforts at research universities.
But irrational and hasty caps to indirects put all of that in jeopardy and my heart breaks thinking about the chilling effect this is going to have on scientific innovation if this policy stands.
###
If you feel the same way, please contact your representatives in congress and explain to them why they must reverse this irresponsible new policy.
A re-analysis of rare disease patients' genetic data delivers 500 new diagnoses
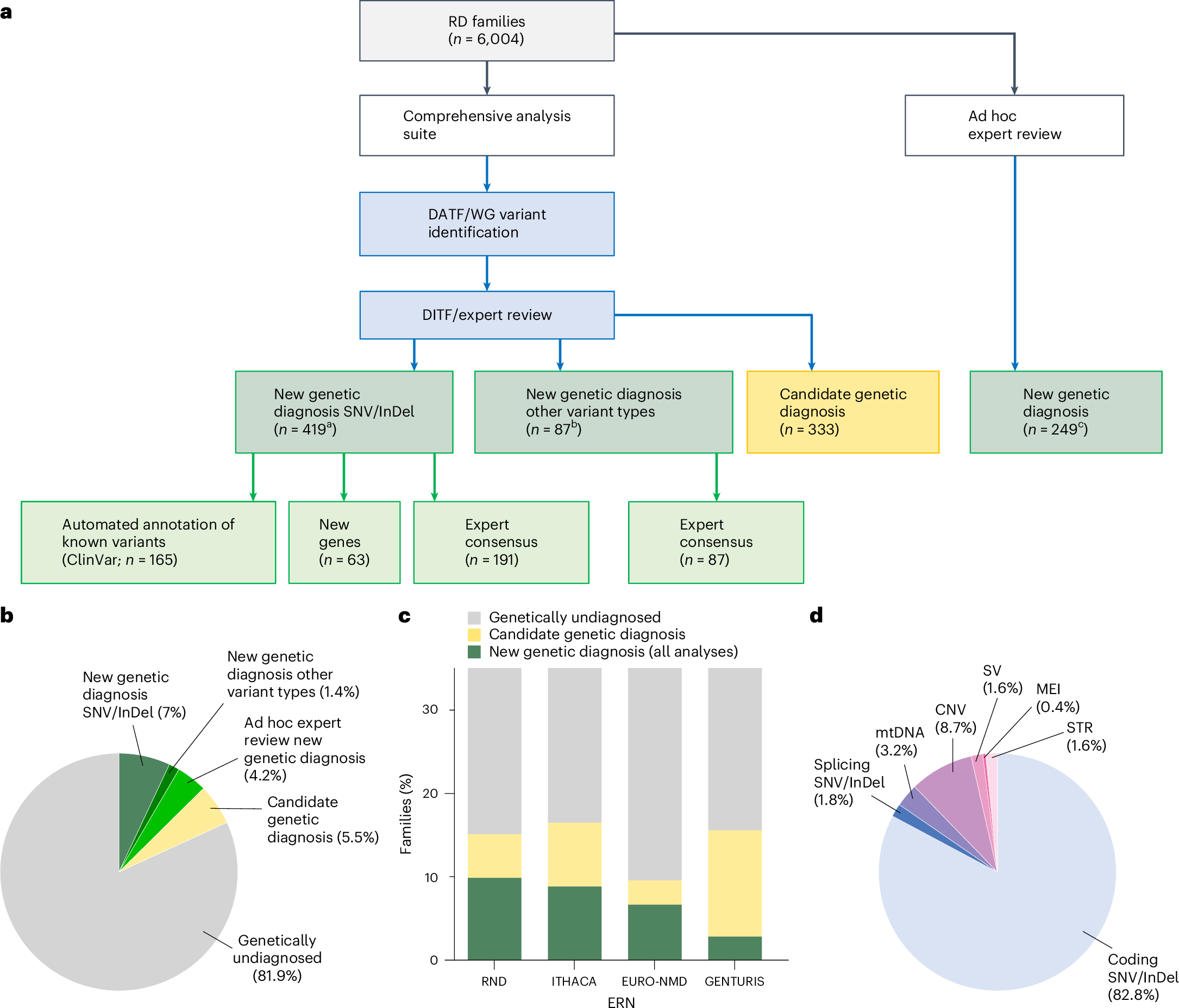
We've heard a lot recently about the utility of DNA sequencing in diagnosing patients with rare diseases.
And, thankfully, whole exome and whole genome sequencing are becoming the standard of care when a patient presents with a likely genetic disease.
The challenge here that still remains is that the diagnostic yield of sequencing a patient can vary greatly - from 20%-70%.
Some of this comes down to the techniques used, because not all sequencing is created equally!
Exomes target the coding regions of the genome (that aren't GC rich and don't have repeats), Short-Read whole genomes capture all of the variation that occurs in coding and non-coding regions but struggles to capture large genomic rearrangements (Inversions and translocations in particular, or mutations that happen in pseudogenes), and while long-reads provide the most comprehensive option for picking up genomic insults, they're still pretty pricey and just barely making their way into the clinic!
But the other issue at hand here when it comes to the variability we see in the solve rate is that when we sequence DNA, we find things, A LOT of things.
For example, you and I have, on average, 5 million single nucleotide polymorphisms (SNPs), 600,000 small insertions and deletions (indels), and 25,000 structural variants in our genomes.
Of these, a few hundred will be "damaging" with unknown effects.
And the same is true for people who are afflicted with a likely genetic disease!
That is to say, finding a genetic cause for a disease really is like trying to find a needle in a haystack!
This also means that sometimes we miss disease causal mutations the first time we go digging through someone's genetic data and as informatics techniques improve, and we learn more about what mutations are associated with disease, we can go back and reanalyze old genomes to find new causes!
That's exactly what happened in this week's paper where researchers and clinicians from the Solve Rare Disease consortium (Solve-RD) re-reviewed genetic data from ~6,000 patients and 3,000 of their family members to come up with new diagnoses.
A summary of the results can be seen in the figure above where a) shows a breakdown of how the new genetic diagnoses were obtained b) a pie chart showing the diagnostic yield of this latest effort in the undiagnosed population c) bar chart showing yield per disease area: RND - rare neurological diseases, ITHACA - intellectual disability, telehealth and congenital anomalies, EURO-NMD - neuromuscular diseases, GENTURIS - genetic tumor risk syndromes d) Pie chart showing the breakdown of the disease causal variant classes
Overall, the re-analysis was able to provide a new diagnosis to 506 patients.
The hope here is that as we learn more, future analyses within this cohort will be able to turn up even more solves.
But since 95% of the patients in this analysis were exome sequenced and only 1.6% of the solves were from structural variants, maybe this cohort of undiagnosed patients would benefit from sequencing with more comprehensive sequencing methodologies that are better able to capture non-coding variation and structural variants.
###
Laurie S, et al. 2025. Genomic reanalysis of a pan-European rare-disease resource yields new diagnoses. Nature Medicine. DOI: 10.1038/s41591-024-03420-w
Formalin-fixed, paraffin-embedded (FFPE) tissues in oncology diagnostics: is there a better way?

But, before we get to that, what is FFPE?
Formalin-fixation was discovered as a cellular preservation technique in the late 19th century and quickly became the most common embalming method throughout the 20th century.
While useful in preserving dead people and animals, it's also quite good at preserving tissues for scientific analysis because it chemically 'cross-links' proteins together making them much harder to degrade.
However, to prevent tissues from shriveling up and losing their morphology, the preservation technique requires a physical stabilizer.
Enter paraffin wax.
After formalin-fixation completes (~24hr), the tissue is embedded in a wax block.
This is followed by 'sectioning' with a microtome (cuts razor thin slices), and finally mounting the tissue sections on a microscope slide for viewing.
This process is great for understanding cellular pathology, but terrible for molecular analysis because getting all of the good bits out of an FFPE sample is very challenging.
Pros:
Preservation of cellular morphology and proteins for imaging studies
Cons:
Everything else
Formalin damages nucleic acids: the fixation process causes DNA and RNA to fragment and this degradation can continue even during cold storage. This can result in a significant reduction in signal, which is important in samples with low tumor fractions.
Fixation can introduce base modifications: deamination of cytosine to uracil and transitions (A>G,T>C) are most common. Base modifications are never ideal because they can result in the reporting of false positive results.
Loss of long range information: because formalin fixation breaks nucleic acid, long range sequence context is lost. Meaning, it's harder to detect gene fusions or chromosomal anomalies using traditional sequencing methods.
Sequence chimeras are prevalent: the damage caused during fixation creates sequence overhangs or single stranded DNA that ends up recombining to create new 'chimeric' sequence that didn't exist in the original sample.
De-paraffinizing a sample is not fun: this involves xylene or intense sample sonication with specialized equipment. One method is unpleasant, the other is expensive.
Treatment with enzymes during processing can reverse some of the damage, but they don't work miracles.
Fortunately, there are alternatives to FFPE and because we know formalin damages nucleic acid, we've been able to develop new methods that are more gentle on the molecular components we're most interested in testing.
These include:
Processing fresh tissue
Flash freezing in liquid nitrogen
Preservation in a tissue stabilizer solution like AllProtect or RNAlater
However, the biggest hurdle is updating the clinical workflow to add the collection of a molecular friendly specimen (in addition to the ones that end up in the jars of formalin!)
Biologically female mammals have two X chromosomes, but what might surprise you is that one of those X's is turned off.
The first person to recognize something different about female animal cells was Murray Barr.
In 1948, he discovered in cats that female cells have a 'nucleolar satellite' which appeared as a black dot in the nucleus.
This was odd because a similar structure could not be found in males.
It was hypothesized that this 'satellite' material was related to sex chromosomes and it later became known as the 'Barr Body.'
Over the span of 11 years, the Barr Body was used to easily discriminate the biological gender of cells.
But in 1959, Susumu Ohno showed that the Barr body was actually a condensed X chromosome!
Two years later, Lyonization, or X-inactivation, was first proposed by Mary Lyon after she observed a correlation between which X ended up being condensed and the coat color of female mice.
Ok, that’s great, but why’s it important for one of the X chromosomes to be turned off?
Dosage.
This is a fundamental concept in genetics and gene duplications or deletions can increase or decrease how many copies of each gene are in the genome.
Issues with dosage are most apparent in diseases like Down Syndrome where individuals have an extra copy of chromosome 21.
This imbalance results in the production of more protein than is required for normal cellular function and also is responsible for the observed phenotypes.
So, X-inactivation ensures that just the right amount of protein is produced from the code written in the X chromosomes!
While genetics is good at correlating genetic changes with broader phenotypes, it can’t prove a direct relationship.
And so the molecular sleuthing began in 1991 with Huntington Willard and his team who located the 'X inactivation center' and discovered a long non-coding RNA (lncRNA) that seemed to encapsulate and condense the inactivated X chromosome.
They named this lncRNA the X-inactive specific transcript or, XIST, for short.
But there was still a nagging question about whether this actually turned off gene expression.
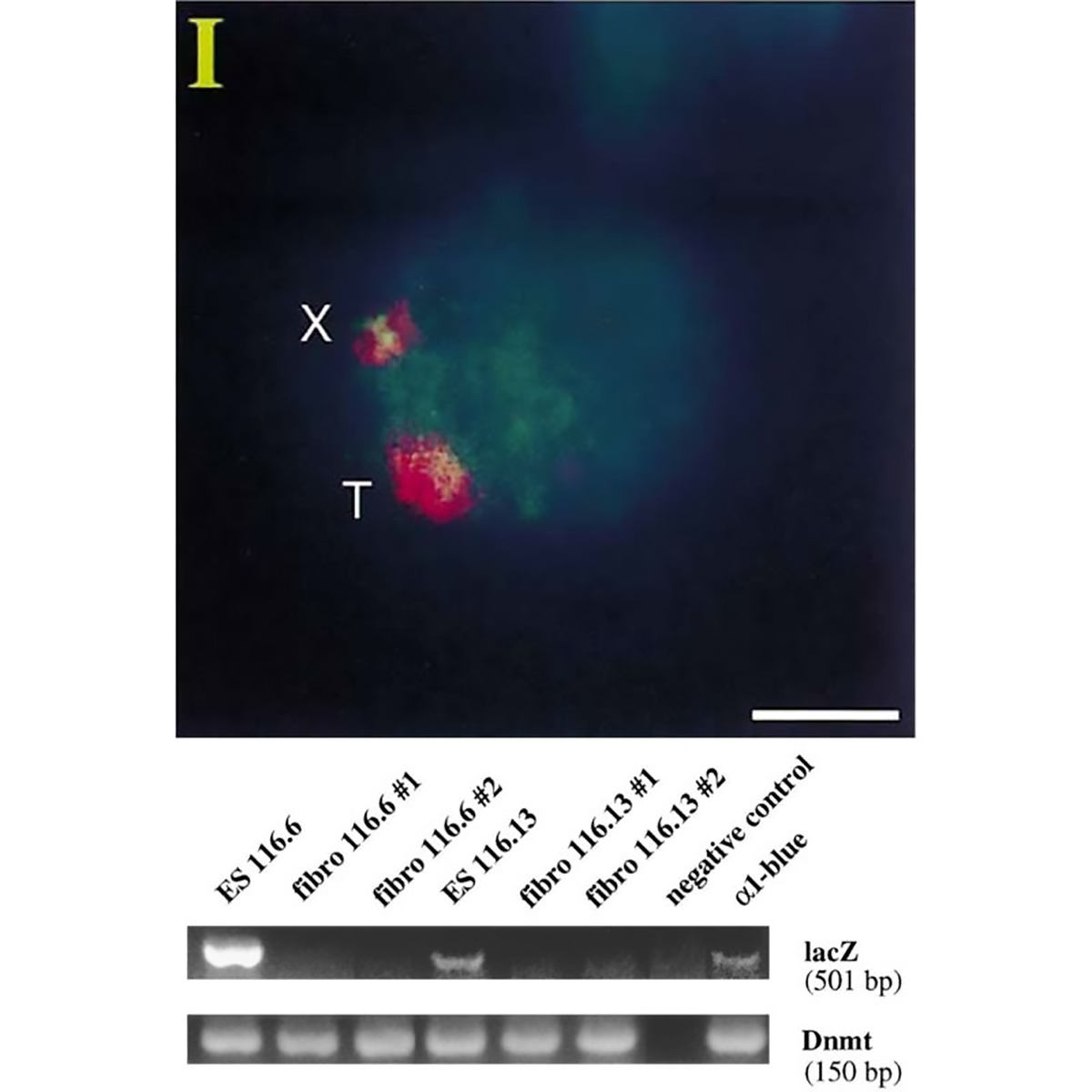
The figure above is the work of Jeannie Lee, who, in 1996, was a postdoc in Rudolf Jaenisch’s lab.
Lee copied the XIST sequence into a yeast artificial chromosome (YAC) that expressed a common ‘reporter’ gene, LacZ.
When introduced into mice, this YAC (T) and the mouse X chromosome (X) both expressed XIST RNA (red cloud).
But the proof that XIST represses gene expression came when looking at LacZ.
The gel shows that LacZ is expressed in embryonic stem cells (Lanes 1 and 4) but not in differentiated fibroblasts (Lanes 2,3,5 and 6)!
This work represents the first direct evidence of a functional role for XIST and its broader involvement in repressing gene expression from the inactivated X chromosome.
###
Lee JT, et al. 1996. A 450kb Transgene Displays Properties of the Mammalian X-Inactivation Center. Cell. DOI: 10.1016/s0092-8674(00)80079-3
Were you forwarded this newsletter?
LOVE IT.
If you liked what you read, consider signing up for your own subscription here: