Omic.ly Weekly 54
December 16, 2024
Hey There!
Thanks for spending part of your week with Omic.ly!
This Week's Headlines
1) Expert panel on clinical microbiome use: "More evidence needed"
2) There are lots of tools for probing the epigenome
3) Automated sanger sequencing's fluorescent beginnings
Here's what you missed in this week's Premium Edition:
HOT TAKE: Like a wounded gorilla, Illumina is roaring back to life with hopes of turning their negative growth numbers into positive ones
Or if you already have a premium sub:
An international panel says clinical microbiomes aren't ready for primetime...yet
Microbiome testing has emerged in the last few years as one of the hottest new wellness trends.
And companies have sprung up that claim to be able to screen the microbes in your gut and provide recommendations for curing everything from food sensitivities to cancer.
I think most of us are aware that the majority of what these companies are promising is a fantasy.
And that's because the microbiome and our understanding of it are still in the earliest stages of development.
There's no question that the microbiome is important with respect to gut and immune health, metabolism, and our response to drugs, but just because we can test for something doesn't mean we understand it.
Nor is all of the data that is generated on this topic good data!
But that hasn't stopped companies from offering these services and providing guidance to consumers on how to use the results to hack their microbiomes.
Fortunately, there are those among us who approach the microbiome more seriously and understand that while we may not know much about the microbiome right now, there will likely be clinical diagnostic applications for it in the future.
What that means, though, is that the clinical community needs to come to a an agreement on how to perform and interpret microbiome testing in clinical practice.
That's why it was really interesting to see a consensus statement published a few weeks ago on the use of clinical microbiomes.
It was compiled by an expert panel of 69 microbiome researchers from 18 countries who see the potential of microbiome applications in the clinic but who worry about the current implementation of available tests.
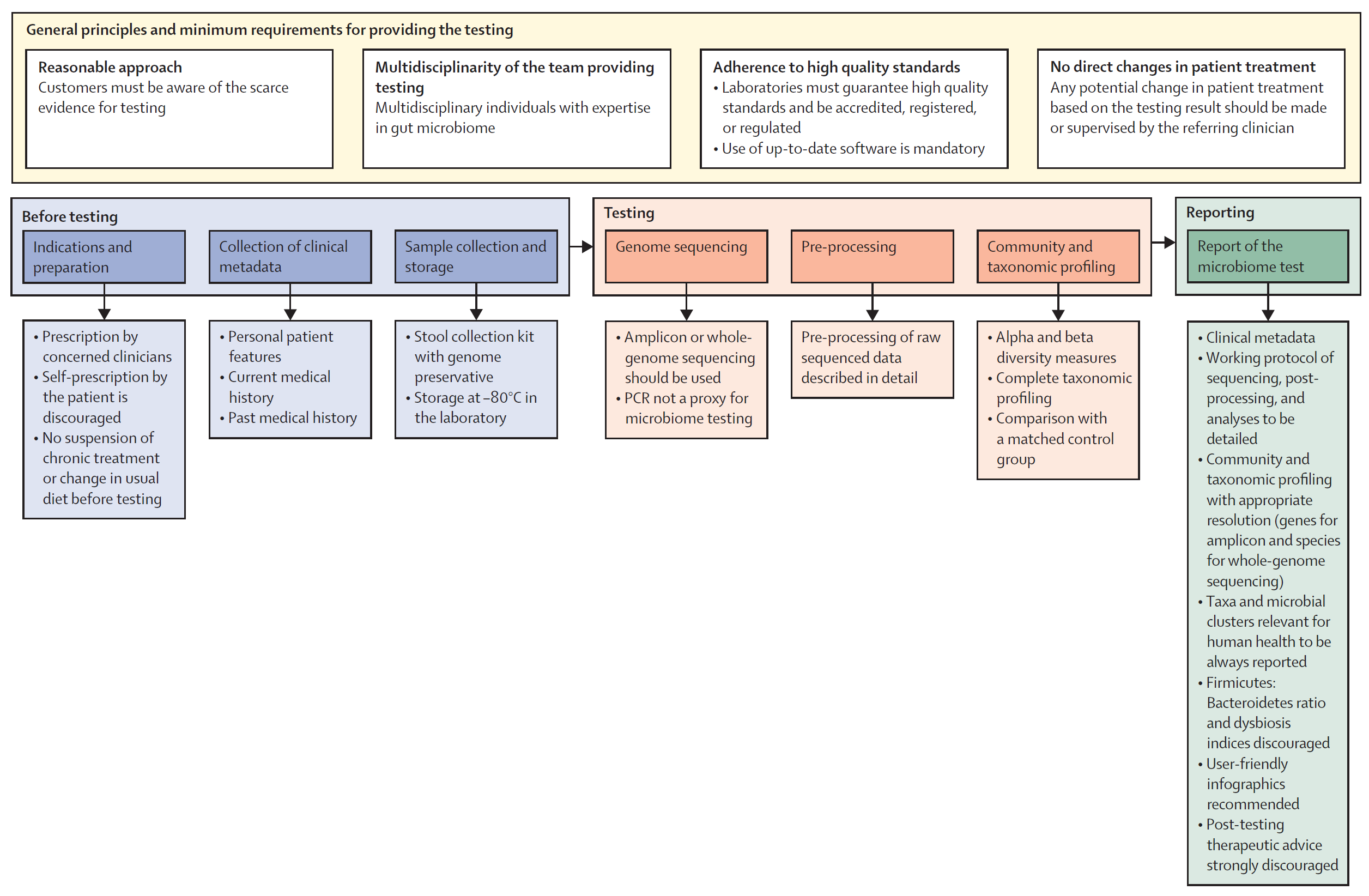
In the statement, they outline 5 key areas of importance (which I'll dig into) and provide a framework (Figure above) for implementing a microbiome testing program.
1. General principles and minimum requirements for providing diagnostic microbiome testing
The panel agrees that anyone prescribing a microbiome test should be upfront about the clinical and scientific validity of the test - specifically highlighting that there's limited evidence for the use of microbiome testing in the clinic. Since microbiomes are so complex, providers should have clinical teams with diverse backgrounds who understand the technology used and how to apply the results in clinical practice. The labs performing the testing should be of high quality, be accredited, regulated, use up-to-date bioinformatic pipelines, protect patient data and use non-proprietary laboratory and analysis methods that can be externally validated (🤔).
2. Procedural steps before testing
Since there is little clinical evidence for the use of microbiome testing, the intended use of the test must be clear and testing should only be performed under clinical guidance (no patient requests!). Essential clinical information such as "age, gender, BMI, dietary habits, smoking and alcohol status, gut transit time, comorbidities and medications, and past medical history" should be collected prior to testing to inform a proper diagnosis. Samples should be collected in a way that avoids contamination, they must be stabilized with a preservative, and stored appropriately at the lab (-80 C) prior to testing.
3. Microbiome analysis
Microbiome testing must be done by either amplicon sequencing (16S) or whole genome sequencing. PCR and culture based methods are not appropriate for getting a complete picture of a microbiome. Analysis pipelines must be well documented and report alpha and beta diversity along with community composition (as a percent). The analysis should also include a comparison to matched controls and, in specific clinical scenarios, longitudinal (sampling over time) may be appropriate to inform a diagnosis.
4. Characteristics of reports
The panel suggests that reports look like standard reports including a brief overview of the test method. Microbiome specific things to include are: alpha and beta diversity, community composition down to the deepest taxonomic resolution (phylum, genus, species, strain, etc...), and taxanomic shifts observed over time or compared to healthy matched controls. The panel discourages the reporting of a dysbiosis index, Firmicutes-to-Bacteroidetes ratio, "healthy" reference ranges, or any therapeutic advice (this should only come from a physician).
5. Relevance of microbiome testing in clinical practice: present and future
If it wasn't immediately obvious, this expert panel does not see much use for microbiome testing in the clinic except for in a few key scenarios (although, even in those ie H. pylori infection, they don't believe microbiome testing is strictly required...), but they do not discount the future potential of such testing and suggest that ongoing research on the microbiome's association with diseases is important. In the meantime, the panel suggests training clinicians on the basics of microbiome science and report interpretation to prepare for a future when the clinical evidence supports broader use of this type of testing.
###
Porcari S, et al. 2024. International consensus statement on microbiome testing in clinical practice. The Lancet Gastroenterology & Hepatology. DOI: 10.1016/S2468-1253(24)00311-X
If epigenetics is the study of modifications that aren't coded in the DNA, how can we use DNA sequencing to learn anything about the epigenome?

Epigenetics seeks to understand how non-sequence elements regulate gene expression.
This control occurs as a result of many factors including the external environment, time of day, temperature, stress, location of a particular cell within a tissue, etc.
All of these factors can change what parts of the DNA can be accessed for conversion into functional proteins!
And, this control occurs mainly at two levels:
1. Methylation of Cytosines (5mc) in DNA can make a region unrecognizable.
2. Modification of histones with methylation, phosphorylation or acetylation can result in the formation of heterochromatin (closed access) or euchromatin (open access).
How can we get at this information?
With sequencing!
Cytosine Methylation:
Caveat! PCR amplification of DNA results in the removal of methylated Cytosine - it’s recognized as just plain old cytosine by polymerases, so in most cases you have to pre-treat the DNA to detect it!
1. Bisulfite Conversion - Bisulfite exposure converts unmethylated Cytosines to Uracils. Subsequent PCR amplification during library creation converts those Uracils to Thymine, and in the resulting sequencing data, any position the remains as a Cytosine was methylated in the original DNA strand!
2. Affinity Enrichment - Certain proteins bind specifically to 5mc. You can affinity enrich these proteins using capture techniques. Another alternative is to use a capture based technology where bisulfite converted DNA is bound to RNA probes designed to specifically pick up regions of known methylation.
3. Direct 5mc Sequencing - Both PacBio and Oxford Nanopore can detect 5mc directly, spitting out 5-bases with high accuracy (A, C, T, G, and 5mc)!
Histone Modifications:
1. ChIP-Seq/CUT&(RUN/TAG) - All of these use antibodies that recognize specific histone modifications like acetylation and methylation to map these marks throughout the genome by sequencing the DNA that they're stuck to!
Chromatin Accessibility:
1. DNase-I Hypersensitivity/ATAC-seq - Euchromatin, the open kind, can be chopped up by enzymes that cut DNA while heterochromatin, the closed kind, protects DNA from this treatment. Sequencing the fragments that fall in the 200-500bp size range tells you where there's open chromatin!
2. Hi-C/Looping - Certain proteins, like CTCF, create 'loops' in chromatin that can activate or repress gene expression. These can be mapped using various techniques like Hi-C which ligates together fragments that are 'close' to one another.
But, epigenetics is complicated, and these marks can differ widely across tissues, cell types, and populations of cells!
They can also change frequently...
So, what's the point?
Epigenetic dysfunction, among other things, can cause rare diseases, cancer, or affect how we age.
But that's a discussion for next week!
We ‘completed’ the human genome in 2003, thanks mostly to commercial industrial engineering.
Genome sequencing didn’t really enter the household vernacular until the announcement of the Human Genome Project in the early 90’s, and that was only possible because of major advancements in sequencing technology.
For the better part of a decade, Sanger sequencing was performed using radiolabeled nucleotides and chain terminators, one for each base, requiring a lane for each A, C, T, and G.
In 1987, George Trainor and a team at DuPont got the bright idea, literally, to use fluorescently labeled chain-terminators, enabling the multiplexing of the Sanger sequencing reaction into a single tube.
This might sound like a simple and obvious feat, but it wasn’t!
The advancement here isn’t just “add fluorescently tagged bases and BOOM! Here’s the data.”
They had to find a compatible polymerase, synthesize the dyes, tag the nucleotides, and engineer an optical system for data acquisition.
The system itself is an engineering wonder: there’s a laser, a beam expander, a suite of scanning optics, two detectors with their own filter stacks, all aligned to excite and detect the dyes in the sequencing gel bands as they pass across a laser beam.
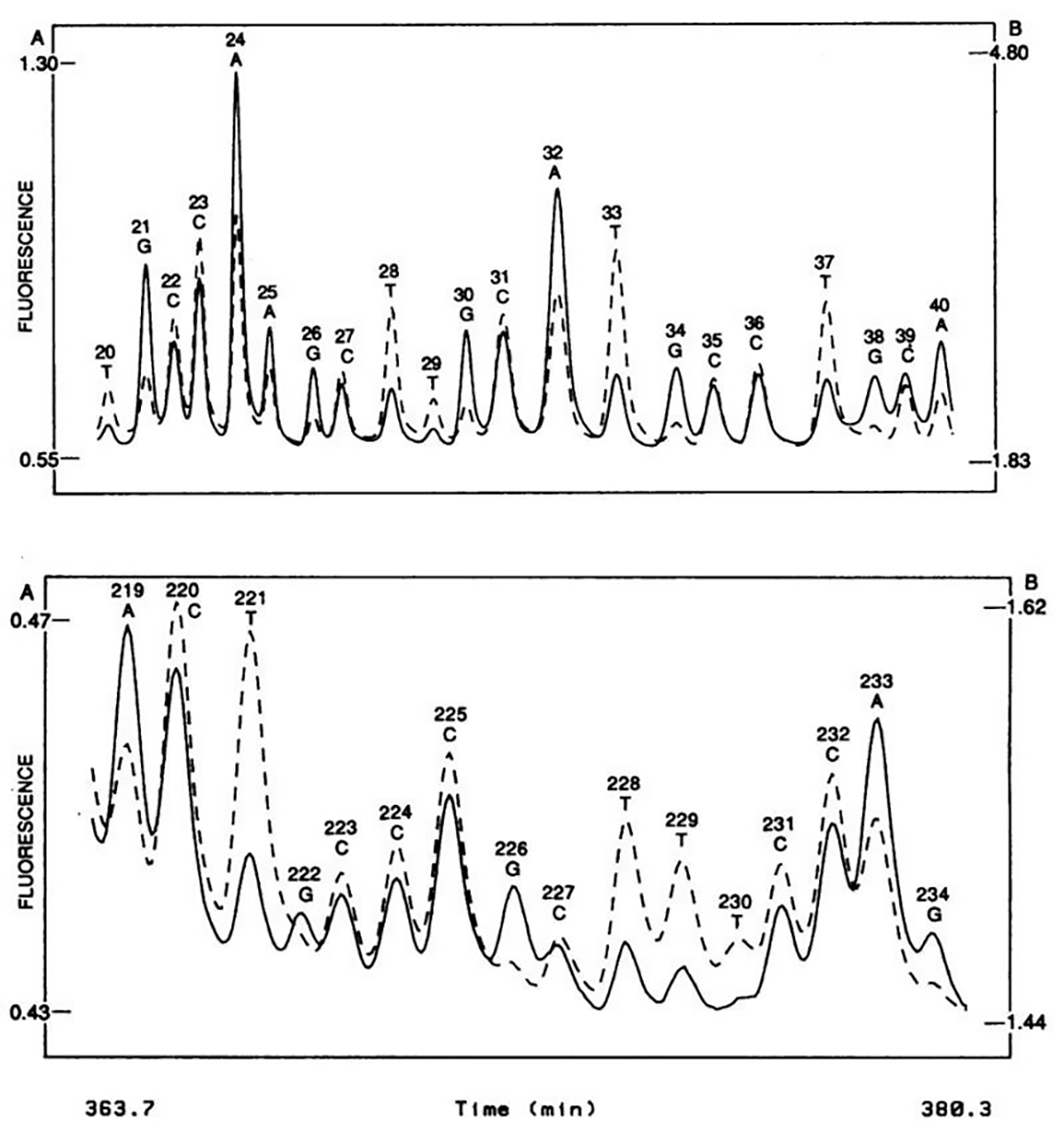
The figure above shows what this groundbreaking system was able to accomplish - the sequence of two 20 base pair stretches of M13mp18, a lac phage vector.
The solid line is the fluorescence intensity observed by the A, G detector and the dashed line the intensity observed by the C, T detector.
These plots should look suspiciously familiar to anyone that’s done fluorescence based Sanger sequencing and it probably isn’t a surprise that, through a licensing agreement, the principles of the technology in this paper made their way into the first four color commercial sequencer in 1988, the ABI 370A.
Despite this process remaining gel based: multiplexing the reaction, eliminating the radioactive nucleotides, ditching the film, and automating the acquisition of the sequencing data were all a huge triumph.
But because it still required the pouring of a gel, it gave grad students and postdocs something to complain about for at least another decade; unfortunately for them, the capillary electrophoresis based ABI Prism line wasn't introduced until 1996.
###
Prober, JM, et al. 1987. A system for rapid DNA sequencing with fluorescent chain-terminating dideoxynucleotides. Science. DOI: 10.1126/science.2443975
Were you forwarded this newsletter?
LOVE IT.
If you liked what you read, consider signing up for your own subscription here: