Omic.ly Weekly 51
November 25, 2024
Hey There!
Thanks for spending part of your week with Omic.ly!
But before we get to this week's juicy bits, I wanted to let you know that this is now a two blog household!
My wife who is an epidemiologist and who spent her grad school days virus hunting H5N1 in Cambodia has also started a website debunking public health misinformation.
If you're interested in following along you can do so on BlueSky, Facebook, or at her website: FactContagion.org
This Week's Headlines
1) Metagenomics hits the clinic to diagnose CNS infections
2) Transcriptomics is better with long-reads
3) Molecular Biology is the offspring of biochemistry and genetics
Here's what you missed in this week's Premium Edition:
HOT TAKE: A new administration isn't going to just make the new FDA rule on laboratory developed tests go away
Or if you already have a premium sub:
Whole genome metagenomics is better than the standard of care in detecting the causal bugs of CNS infections
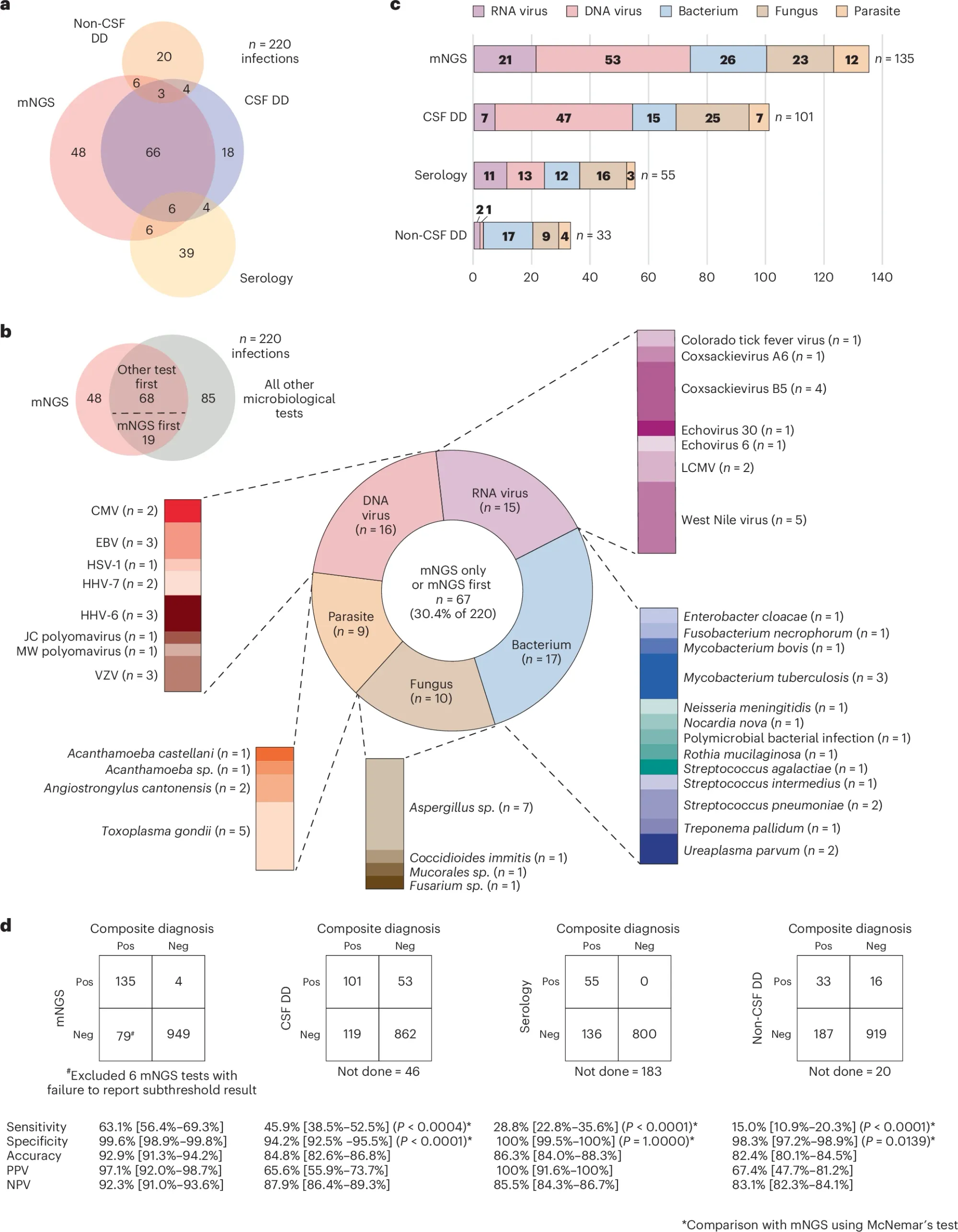
Unfortunately, whole genome metagenomics has struggled to gain a foothold in the clinic.
This is largely because this testing is expensive, and at least in the US, insurance companies refuse to pay for things where there isn’t sufficient clinical evidence to justify those costs.
At least that’s been the excuse they highlight in their refusals to pay for such innovative and life savings tests.
However, new studies, like the one shown in the figure above, will hopefully start reversing those coverage denials.
In diagnostic testing, speed to result is always of critical importance and we often optimize processes to be sure that samples are received and processed in the shortest “turn around time” possible.
This is because every result is a patient, and many of the tests that we perform, especially the ones that diagnose infectious diseases, are used to help prescribe medications.
And in the case of infections, prescribing the right medication at the right time can mean the difference between life or death.
This is a notable problem in infections of the central nervous system (CNS) which can be uniquely challenging.
You’ve most certainly heard of meningitis (inflammation of the membranes around the brain and spinal cord), but there’s also encephalitis (inflammation of the brain), and very rarely there are infections that can cause both of those at the same time: meningoencephalitis!
Now, diagnosing these kinds of infections can be tricky but the standard of care for testing here is serology (use antibodies to detect infectious agents) or direct detection of pathogenic nucleic acids using something like PCR.
The challenge with these types of assays is that you kind of need to know what you’re looking for, and even running a large panel of serology tests or PCR tests can still miss what’s causing the problem!
But we now have high throughput sequencing that can just sequence everything without a doctor having to guess if an infection is viral, bacterial or the result of a parasite!
In this week’s paper, the authors showed 7 years worth of data performing a metagenomic sequencing test (mNGS) for CNS infections and compared its performance to serology and direct detection with other molecular assays.
In the figure above a) shows a Venn diagram of all of the infections diagnosed by each technique b) is a breakdown of everything detected by mNGS c) is a bar chart showing all of the different types of bugs that were detected and d) compares the performance of each test to the composite diagnosis (consensus) and highlights that mNGS outperforms all other testing modalities
However, mNGS wasn’t perfect and had false positives and false negatives which the authors dig into and highlight that in many of those cases where there were false negatives that host background was high.
One of the biggest challenges in doing metagenomics is separating the host DNA from the infectious signal.
Here the authors used antibodies to remove methylated human DNA from the samples before processing them, but that process isn’t perfect (and not ALL human DNA can be removed this way!)
So, having too much human DNA around can limit your ability to detect the rarer sequences from the infectious agents!
But at the end of the day, it all comes down to performance and mNGS beat out both serology and direct detection testing which makes a strong case for its use diagnosing critical CNS infections in the clinic.
###
Benoit P, et al. 2024. Seven-year performance of a clinical metagenomic next-generation sequencing test for diagnosis of central nervous system infections. Nature. DOI: 10.1038/s41591-024-03275-1
Spoiler Alert: The holy grail of transcriptomics is long-reads.

Transcriptomics is the least talked about of the omics in sequencing because, for whatever reason, DNA gets all the fanfare.
But we are able to function as living organisms because of the activities of DNA, RNA and proteins.
While the genome and DNA are the storage form of our genetic material, the transcriptome is made up of all of the RNA messages derived from the genome.
Now, the funny thing about the information in our DNA is that it's split up into sections called introns and exons.
The introns are removed from the final RNA message during a process called splicing.
On average, an exon is 200bp and on average, a fully spliced human RNA message is 2,000bp.
But RNA is tricky.
That splicing process doesn't always happen the same way in every cell or even in every transcript.
So, cells also contain different versions of these messages, called isoforms, that can contain slightly different combinations of exons.
The inclusion or exclusion of an exon can have a dramatic effect on the function of a protein, so it's kind of important to keep tabs on these isoforms, especially as they relate to disease!
So what happens when you try to do transcriptomics using short-read sequencing?
Well, you first start by turning the RNA into DNA, and then you fragment that DNA so you can sequence it using the traditional short-read process that has a maximum read length of 300bp.
"But Brian, didn't you say that the average RNA is 2000bp? Won't you lose information about what isoforms were present if you chop everything up?"
Fantastic observation!
You'll still pick up the splice junctions and get a rough idea of what exons were connected together, which can help in predicting what isoforms were present.
But long reads are definitely better and you'll capture all of those exons as they exist in a single message without having to guess what was where!
A typical transcriptomics experiment with short reads requires 100-200 million reads.
Since long reads are long, you need about 1/10th as many because you're getting the full length transcript.
Historically, long-read transcriptomics has been pricey, but PacBio has commercialized a transcript arraying method that combines 7-ish sequences into a single read with barcodes separating each in the array.
This more efficiently uses their 16kb reads, effectively reducing their costs and putting them on par with short-read based methods.
Another issue to consider around cost here is that the transcriptome actually represents an amplification of the genome.
Some RNAs are expressed A LOT, and so because you're random sampling from these sequences, you'll pick up the really abundant ones and probably miss the rarer ones.
One way to get around this is to deplete all of those highly abundant messages using enzymatic targeting techniques (CRISPR, RNaseH, etc).
Ultimately this makes your long read dollars go much...longer!
The field of molecular biology was born in 1941 through the marriage of genetics and biochemistry.
This, to this day, is not a very happy marriage.
Both sides tolerate one another, but the biochemists lean on function and the geneticists lean on math and statistics.
The progeny, or molecular biologists, fall in the middle, trying to muffle the yelling and screaming of the other two, but this diversity of ideas is actually what makes science great - even if mom and dad fight sometimes!
But, before we dig into the birth of this dysfunctional family, it is important to highlight that in the 1900's, the idea of a 'gene' was an abstract one.
We all know Mendel and his peapods, that traits are inherited and it was thought that this was all controlled by genes, but 'how' any of that worked biologically was essentially a mystery.
George Beadle was fascinated with the 'how' and spent many years in a Drosophila (fruit fly) lab trying to connect pigments to whatever a gene was.
And he rightly hypothesized it involved biochemical pathways.
He realized that approaching the problem from the perspective of the end product, the pigment, was wrong.
And this meant he had to figure out how to break the 'gene' and link it directly to biochemistry.
But doing that in a complex multicellular organism like a fruit fly was going to be impossible so he switched to a much simpler one: Neurospora!
Now, Neurospora, being a fungus, has certain features that made it ideal for this project.
Fungi are haploid (1 copy of the genome per cell, not 2) for a good portion of their life and heritable genetic changes are easy to track in their offspring because you don’t have to worry about any compensatory interference from an alternate working copy!
Beadle realized he could create mutations in fungi and then link those to biochemical processes.
But how do you make mutations in genes in 1941?
With X-rays!
So, he irradiated a bunch of haploid Neurospora and then grew them on different types of food:
One that had everything they needed to live (complete), and one that did not (minimal).
Once he found mutants that could grow on complete but not minimal, he added back individual components to the minimal food to figure out what the fungi couldn't make anymore.
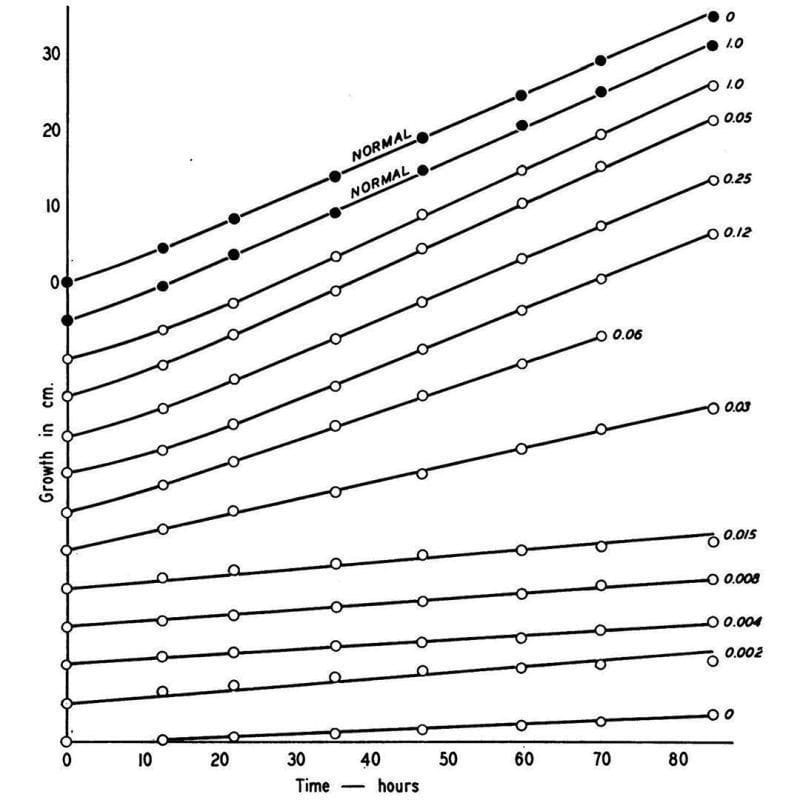
What he found is shown in the figure above, which displays the growth rate of normal Neurospora (black dots) and a mutant (open dots) that couldn’t make vitamin B6 anymore.
This mutant could be saved though, and its growth rate controlled based on how much B6 (numbers) was added back to the food.
Given enough B6, the mutant could grow just as well as the normal one!
This might seem super basic, but it was the first clear evidence that linked a biochemical process directly to a heritable gene - the rest is molecular biology!
###
Beadle GW and Tatum EL. 1941. Genetic control of biochemical reactions in Neurospora. PNAS. DOI: 10.1073/pnas.27.11.499
Were you forwarded this newsletter?
LOVE IT.
If you liked what you read, consider signing up for your own subscription here: