Omic.ly Weekly 46
October 21, 2024
Hey There!
Thanks for spending part of your week with Omic.ly!
Maybe we'll just switch to Monday mornings for the newsletter to post...
My weeks and weekends have been a bit of a hurricane themselves lately but hopefully I can get back to the normal Sunday schedule soon!
Thanks for being patient with my ever changing schedule!
This Week's Headlines
1) Complex genetic traits are getting taken to new heights
2) Don't let highly expressed transcripts get the best of your RNA-seq dataset
3) Solving the DNA double-helix took a village
Here's what you missed in this week's Premium Edition:
HOT TAKE: Element Bioscience makes a big bet on rapid exome sequencing
Or if you already have a premium subscription:
The genetics of height gets the whole genome treatment
Genetics can get complicated quickly, and that’s no secret when we’re talking about the genetics of height.
Sometimes we scientists try to over-simplify things in genetics.
We talk about mutations and variants like there’s a strict cause and effect relationship between them and some disease or phenotype.
This isn’t true for, like, most things - genetics is complicated as h e 🏒 🏒 more often than it’s not!
Even for things like Mendelian traits and diseases (where there is a clear gene-phenotype effect) there’s still variability in penetrance for most mutations.
This simply means that with respect to diseases, mutations can show variability in their severity, if they even result in disease at all.
But this ‘simple’ situation gets way more complex with *drum roll* complex diseases and traits!
Things like eye color and height are considered to be complex traits, meaning that they are controlled by the activities of multiple genes and/or environmental factors and do not typically follow Mendelian inheritance patterns.
Geneticists also refer to complex traits as being polygenic.
And for the longest time, most research done on complex traits used array genotyping to try to map all of the variants or regions in the genome that contributed to their presentation using genome wide association studies (GWAS).
In the example of eye color, we’re talking about what regions of the genome control color or shade, and with height we’re talking about…height or how tall someone is.
But complex traits get even more complex than just the mutations associated with the coding regions of multiple genes.
We’ve also learned in the years since our initial sequencing of the human genome that non-coding variants (things in enhancers, promoters or intergenic regions) can also play important roles in how genes are expressed!
Which means that for us to totally understand complex traits like height, we need to look at all variants in the genome that can contribute to a phenotypic effect.
Unfortunately, in our years of doing GWAS, we focused mainly on common variants in human populations that contributed to these traits which means we’ve missed a good chunk of the variation that contributes to the phenotypes that we see.
For height, it’s estimated we’re still missing 50% of that variation!
However, as the cost of sequencing has declined, that means we can stop just looking at common variants, and start using whole genome sequencing to look at rarer ones to help find that missing variation.
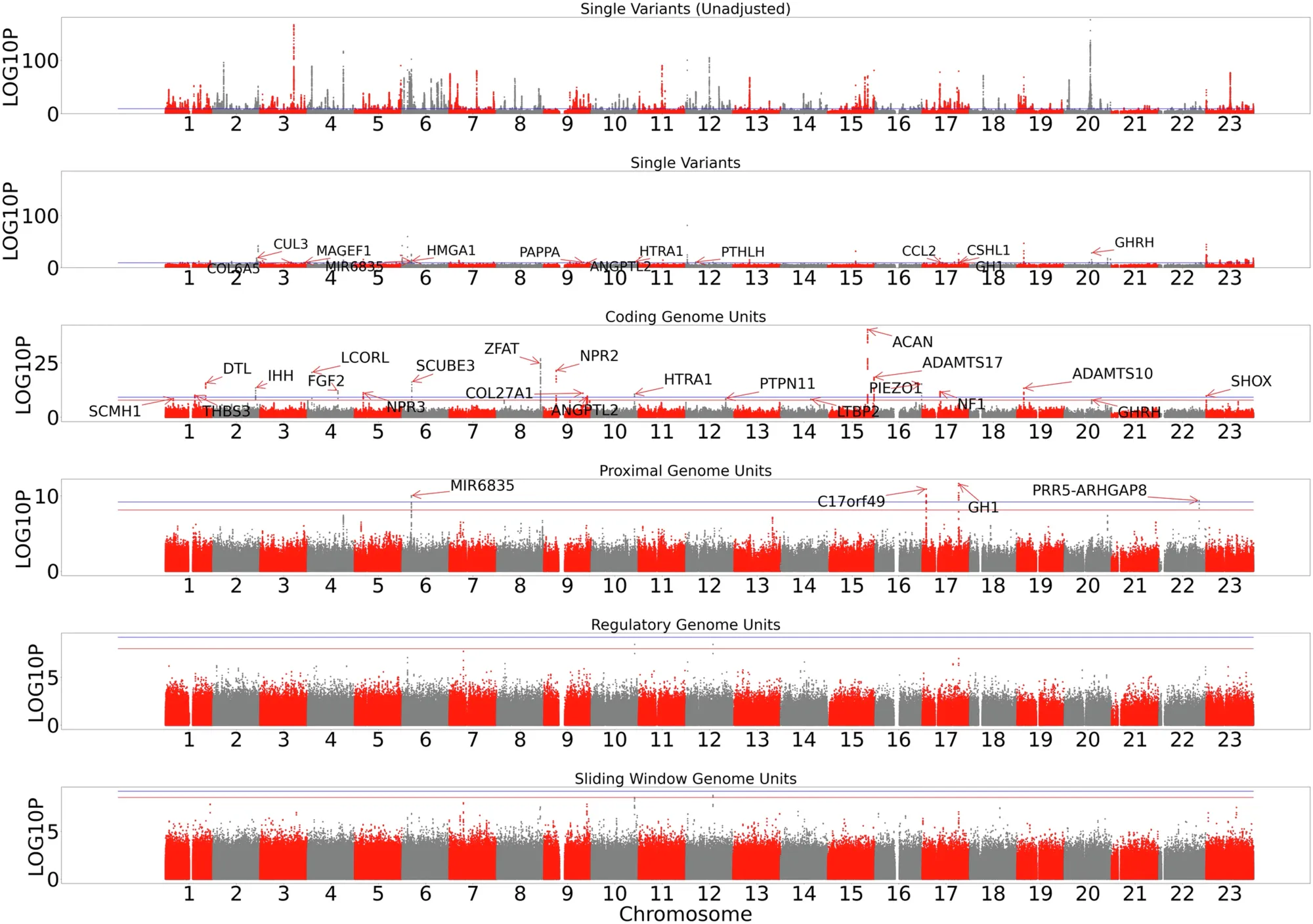
And that’s what can be seen in the figure above.
The researchers used whole genome data from the UK Biobank, All of Us, and TOPmed to suss out new genes and non-coding regions that control the variability we see in height.
The figure shows Manhattan plots for single variants, genes, proximal variants (within 5kb of a 5/3’ UTR), regulatory variants (more than 5kb from a gene), and whole genome sliding window (large regional contributions).
Locations were panel significant if they crossed the red line, or study wide significant if they crossed the blue line.
They found new single nucleotide variants or aggregate proximal variants associated with height in HMGA1, C17orf49, GH1, CSHL1, PRR5-ARGHGAP8 and MIR6835.
This study highlights the importance of considering the contributions of rare variants in complex traits for future genome wide association studies.
###
Hawkes G, at al. 2024. Whole-genome sequencing in 333,100 individuals reveals rare non-coding single variant and aggregate associations with height. Nature Communications. DOI: 10.1038/s41467-024-52579-w
Whole transcriptome sequencing AKA RNA-seq: The good, the bad, the dynamic range?

DNA is the storage form of our genetic information, but transcription is the process in which DNA is converted into the message, the ribonucleic acid (RNA), that codes for proteins!
During transcription, the double helix is unwound and the DNA is bound by a bunch of proteins called transcription factors that recruit an RNA polymerase to begin the conversion process.
The polymerase uses DNA as a template to create the complementary RNA message by attaching RNA bases together.
That's A, C, G, and U!
"Me?"
No, Uracil.
RNA is special, unlike DNA, it doesn't have T or Thymine, it has Uracil which is really just a Thymine that's missing a methyl group.
As the polymerase chugs along it copies the exons (the parts that code for protein) and the introns (the parts that don't code for protein) into a single strand of RNA. The introns are removed during a process called splicing where sequences in the introns and at the ends of exons are recognized by a protein complex called the spliceosome. Ultimately all of the exons are spliced together to create the final RNA message.
This all happens in the nucleus of the cell, but the conversion of that RNA message to functional protein happens in the cytoplasm and on the endoplasmic reticulum (the cabbage-y outer shell of the nucleus!) Here the RNA is bound by another protein complex called the ribosome, and this complex reads the RNA message to create proteins from amino acid building blocks (this is called Translation).
Why did I go through all of the effort to tell you this?
Because, each cell only has two copies of DNA, but that DNA can be turned into thousands of copies of RNA as a result of transcription!
AND all of those RNA messages together make up the transcriptome.
But this also means that RNA is actually a reasonable read out of the biological function of a cell and we can learn even more by capturing those molecules and sequencing them!
RNA-seq can tell us which sequences of DNA actually end up in an RNA message, how much of each message is made, and what isoforms of each RNA message are present (splicing can result in multiple different RNAs being generated depending on which exons are included in the final message).
Unfortunately, since all of the cells in our body express slightly different RNAs depending on the function of a cell, we need to sequence a lot of cells to truly understand the transcriptome!
And double unfortunately, because transcription can create a lot of copies of the same RNA, sometimes very abundant messages, like those for ribosomes or hemoglobin, can dominate the resulting sequencing data causing us to miss messages that may have fewer copies present.
Luckily, we can fix this dynamic range issue using ribo depletion/globin reduction protocols to get rid of these overabundant messages and focus more on the unique RNAs that matter most!
History is written by victors, and that statement couldn't be more true than it is in the case of Watson and Crick's 'discovery' of the DNA double helix.
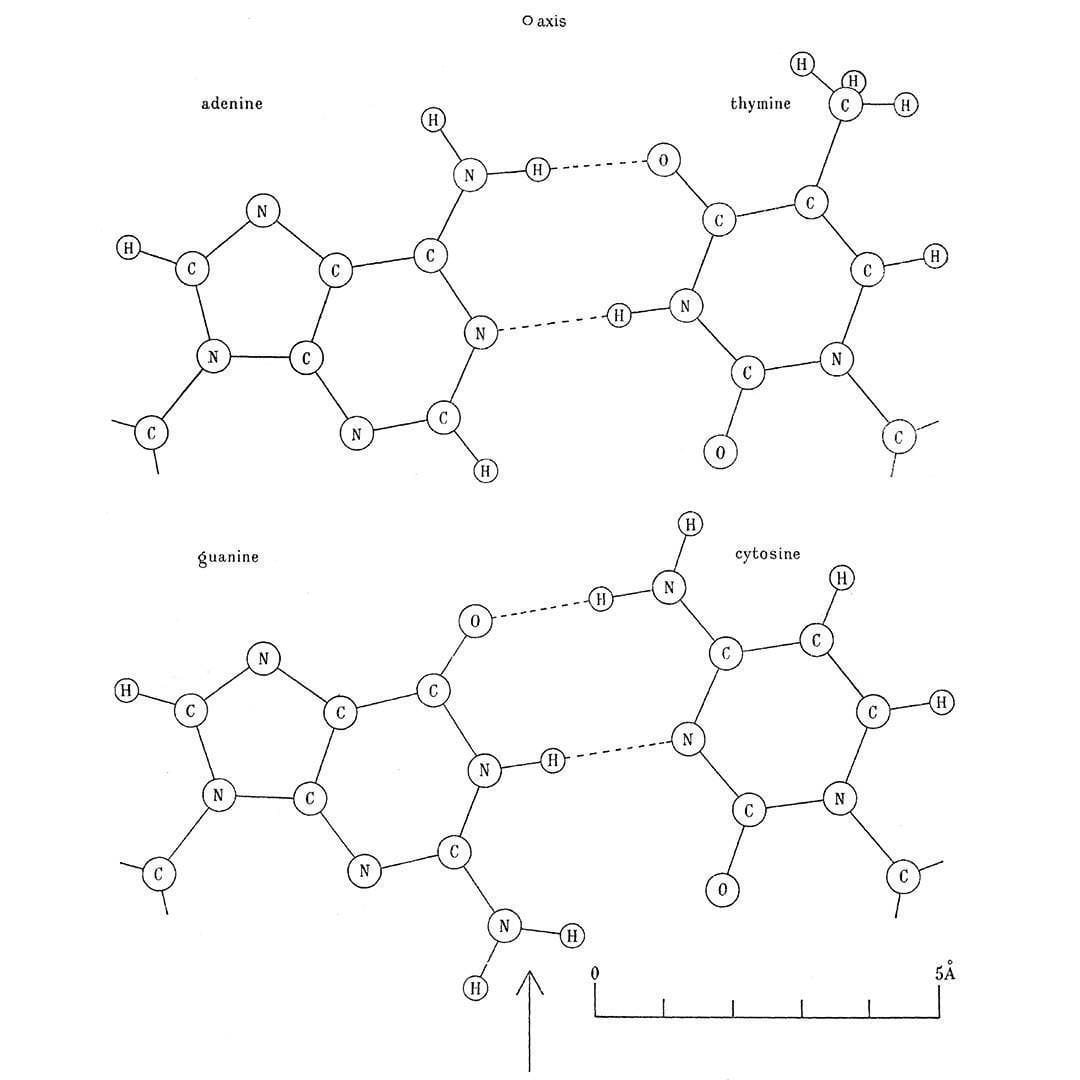
Their structure was published in the April 1953 issue of Nature along with two other papers on the same topic from Wilkins and Rosalind Franklin.
Although they don't cite Franklin in their 1953 paper, they definitely used her data.
It was Franklin's paper that included Raymond Gosling's pristine diffraction of B-DNA, known as Photo 51, that showed the structure of DNA is helical, it's double stranded, and the bases faced inward with the phosphate backbone on the outside.
While this is a good chunk of what you need to know to put the structure of DNA together, there were a couple of additional missing pieces that the boys from the Cavendish Lab had to source.
What gets lost in all of the popular coverage of this discovery is that Watson and Crick didn't perform any experiments, they aggregated the best science at the time to create their model.
Franklin wasn't the only person they borrowed from.
Phoebus Levene's life was spent studying DNA, he's why we call the bases nucleotides and he showed that DNA has a 5'-3' deoxyribose sugar phosphate backbone but also that the bases are composed of adenine, thymine, guanine, and cytosine.
Erwin Chargaff shared with them what he knew about the ratios of the bases, or Chargaff's rule, which is that A and T, and G and C are found paired in a 1:1 ratio.
He had a famously checkered opinion of the duo and even went as far as to say, "I told them all I knew. If they had heard before about the pairing rules, they concealed it. But as they did not seem to know much about anything, I was not unduly surprised."
Next on the list was hydrogen bonding between the bases. This little known fact was cribbed from the thesis work of a graduate student at the time, June Broomhead (Lindsey), who also proposed all of the possible structures for A, T, G and C.
But knowledge of that final piece of the puzzle came from Jerry Donohue who shared an office at Cambridge with Crick.
He noticed that Crick was trying to pair up the bases using their 'enol' forms and so Donohue suggested, based on Broomhead's work, that Crick should try to smash the 'keto' forms together instead because they were much more common.
The figure above is Crick's smashing result: Watson-Crick(-Broomhead-Donohue?) base-pairing. It was published in 1954 in a much longer and more detailed follow-up paper on the structure of DNA.
While historic, this story is nuanced, and I'll leave you with Crick's measured interpretation of the situation.
"What, then, do Jim Watson and I deserve credit for? The major credit I think [we] deserve … is for selecting the right problem and sticking to it."
I tend to agree.
###
Crick FHC, Watson JD. 1954. The Complementary Structure of Deoxyribonucleic Acid. Proc. R. Soc. A. DOI:10.1098/rspa.1954.0101
Were you forwarded this newsletter?
LOVE IT.
If you liked what you read, consider signing up for your own subscription here: