Omic.ly Weekly 41
September 8, 2024
Hey There!
Thanks for spending part of your Sunday with Omic.ly!
This Week's Headlines
1) Let the Nanopore Wars begin: BGI has a new nanopore sequencer
2) Low frequency variants are hard to detect, but don't let that stop you from implying you can catch them all
3) Who discovered PCR first? The answer might surprise you
Here's what you missed in this week's Premium Edition:
HOT TAKE: CRISPR isn't the only relevant genome engineering technique, can we talk about the others too, please?
Or if you already have a Premium Subscription:
The Nanopore Wars are coming and BGI just fired the first salvo in what is sure to be an exciting conflict
Nanopores as sequencers have been a genomics dream since at least the late 1980s.
But, the first patent for this style of sequencer was filed in 2008 by Mark Akeson, David Deamer, William Dunbar, Noah Wilson, and Kathy Lieberman at UCSD.
Deamer had originally thought of a ‘channel’ based DNA sequencer that determined the sequence of bases based on how they disrupted an electrical current during a road trip in 1989 and scribbled his thoughts in a notebook.
These notes would serve as the basis for this patent 20 years later.
The journey to that filing obviously took some thought and experimentation.
A meeting between Deamer and his old friend, Daniel Branton, in 1991 reignited his interest in the topic of channel, or pore, based sequencing and the next 17 years were dedicated to the pursuit of figuring out how ‘nanopores’ could be used to make a better DNA sequencer.
The first fruits of this labor were published in PNAS in 1996 where the team was able to show they could pass DNA strands through pores and generate signals.
But it wasn’t until the early 2000’s that the team was able to actually discern nucleotides within those strands.
However, this was good enough evidence that there was potential in nanopores as sequencers and in 2007, Oxford Nanopore Technologies licensed this technology for commercialization.
The rest, as it were, was history.
That might be a little too flippant, though, since significant development effort has gone into converting this proof-of-concept technology into the current product that ONT sells today.
Fortunately (or unfortunately for ONT), since commercialization to a viable sequencing technology has taken over a decade and a half, that means the original patent is set to expire in less than 5 years.
Which is what makes this recent preprint from BGI on their internally developed nanopore technology so interesting.
In it they detail how they have discovered or engineered pores and motor proteins with less than 50% sequence identity to other nanopore proteins.
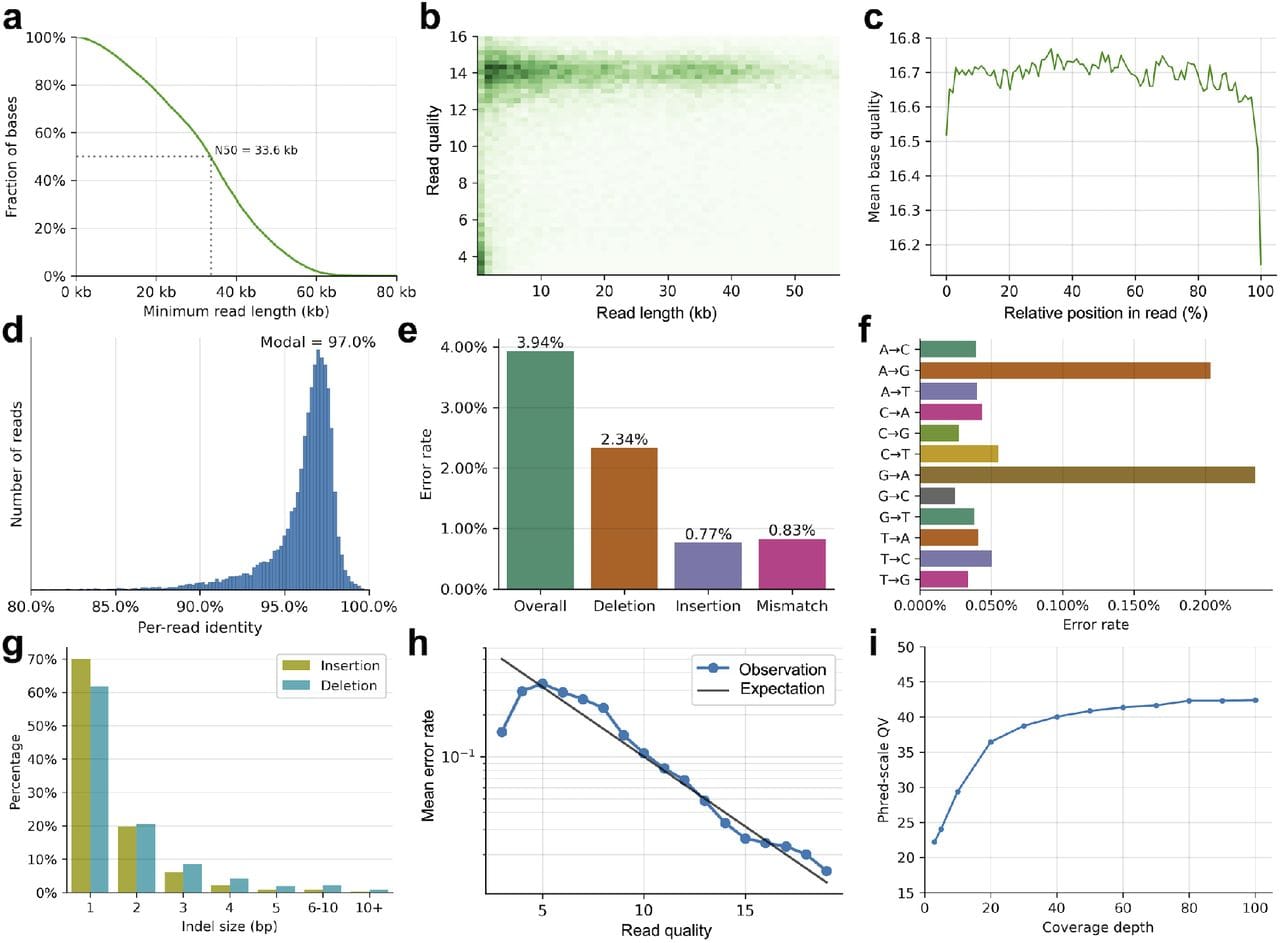
They’ve also packaged this into something that looks and performs similarly to what we’ve all come to expect from nanopores which can be seen in the figure above.
In A) they achieve a read length N50 of 33.6kb (ONT is ~25kb), B,C,D) have a read quality of Q16.7 or ~97% (ONT is Q20+ or >99%), E) Their overall error is dominated by deletions, F) They struggle most with A<>G transitions, and I) their consensus accuracy is on par with currently accepted standards ie >Q30 @ 30x coverage.
Whether this platform struggles with homopolymers (extended runs of the same base) remains to be seen since this is not mentioned anywhere in the paper.
But these results are promising and potentially offer a serious alternative to ONT in the Nanopore sequencing space.
And since they seem to have figured out how to divorce the detection electronics from the pores, they could also offer a significant cost advantage over the competition.
That is if you can wait until 2029 to purchase them in North America.
###
Zhang J, et al. 2024. A single-molecule nanopore sequencing platform. BioArxiv (Preprint). DOI: 10.1101/2024.08.19.608720
Detecting variants can be challenging, especially detecting the ones that aren't very abundant.

Unfortunately, there are a good number of labs that use the default settings for their sequencing analyses, and/or implement premade pipelines that they 'validate' without doing the appropriate amount of work to make sure that the thing they've developed actually performs the way they say it does.
This gets extra tricky in oncology screening where the allele frequencies can drop well below 1% with the latest crop of sequencing based tests advertising sub 0.1% detection capabilities.
But what does it mean to be able to call a heterozygous variant with a 50% frequency in a germline sample or a 0.1% frequency variant in a liquid biopsy?
Can the assay do it every time time?
Can the assay do it in every sequence context?
How do you know?
There are a few really good ways to know, but most places don't do these things because they're not required to:
In silico decimation - this is an informatic technique where the data from a large number of samples is randomly reduced ie take samples with a minimum coverage of 40x at each position and reduce them to 30x, 20x, 10x to see where the assay starts losing the ability to detect specific types of variants. In the case of liquid biopsy samples where the minimum coverage to call a sub 1% variant approaches 10,000x (depending on the quality/error correction strategy), decimation through a much higher coverage range might be warranted. It is also possible to create contrived datasets where variants are randomly inserted into the data at a specific frequency, but these programmatic manipulations of frequencies are only good for evaluating informatics performance, not lab process performance.
Contrived synthetic controls - one way to test process performance is to synthesize a variant into a sequence using a company like Integrated DNA Technologies and then spiking or diluting that sequence fragment into a sample to 'contrive' the mutation. This 'sample' can then be taken through the whole process and be used to determine at what allele frequency the lab process begins to fail to detect the variant (preferably this is done for every gene/target/exon in the panel).
Characterized mixture panels - many companies (and NIST) offer mixture panels. Some are contrived, others are mixtures of cell lines with well known variants, but most importantly, these panels have been independently characterized using sequencing and/or droplet PCR to precisely determine the allele frequencies of the variants contained in the panels. These allow for accurate benchmarking of the performance of an assay using an independent resource.
However, none of these methods is perfect and it's always a good idea for labs to track assay performance post-launch, especially as interesting positive samples are gathered or become available through other sources.
The method below has been cited more than 600,000 times and is one of the most important developments in the history of science.
The polymerase chain reaction (PCR) is used widely to amplify DNA sequences.
Kary Mullis is often given sole credit for developing PCR, which, according to him, he discovered while "riding DNA molecules" during an acid trip in the early 1980's.
But, today's story begins a decade earlier in the lab of Har Gobind Khorana.
Khorana won the Nobel Prize in 1968 for his work figuring out how RNA codes for protein.
The key to his success was that he and his team synthesized their own molecules of RNA, called oligonucleotides.
This allowed them to see what amino acids ended up in proteins after the translation of their sequences.
Khorana continued to focus on scaling this synthesis process to create a full gene sequence.
One of his post-docs, Kjell Kleppe, had an idea to use little pieces of complementary DNA to kickstart these synthesis reactions and copy a target sequence with DNA polymerases.
At the time, it was known that polymerases were involved in copying DNA during cell division and could use a 'primer' to start this reaction.
He presented his method and initial data in 1969 and published a paper where his method of in vitro "DNA repair replication" was described in 1971.
So, why does Mullis get all of the credit for the discovery of PCR?
Mostly because of timing.
In 1970, the Khorana lab was one of the few groups making oligonucleotides.
Kleppe's result was seen as interesting but technically infeasible to scale and the true power of DNA copying wasn't recognized until the early 1980's when cloning and other molecular manipulations were really taking off.
Mullis was employed at that time by Cetus Corporation and he stumbled on the idea of copying DNA using primers and polymerases all on his own.
He published an initial paper on PCR in 1985 using the E. coli ‘Klenow’ fragment (a truncated portion of DNA polymerase) which required manually cycling the reaction in a water bath and adding back enzyme every round because it wasn’t heat stable.
However, Mullis realized he could use the polymerase from a different bacteria, T. aquaticus, which lives in the boiling hot springs of Yellowstone Park.
This polymerase, which we now call Taq, was heat stable and didn't require replacement between cycles!
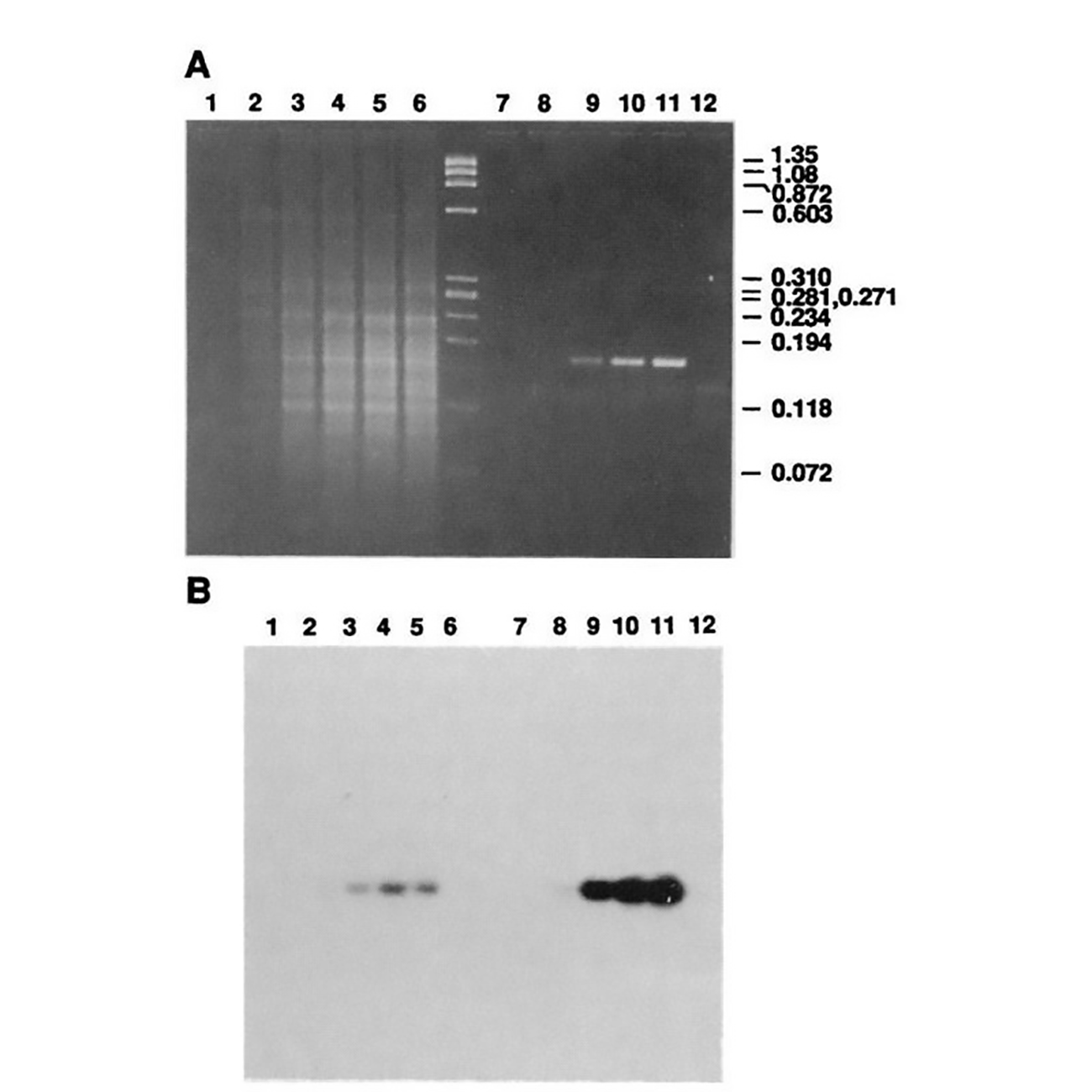
The figure above is Mullis' comparison of Klenow (Lanes 2-5) to Taq (Lanes 8-11).
(A) is an agarose gel and (B) is a southern blot using a radioactive probe to detect the targeted DNA.
The benefits of using Taq are pretty obvious, but its heat stability was game changing because it made the process much cleaner and easy to automate!
Mullis received the Nobel Prize for this work in 1993.
###
Saiki RK, et al. 1988. Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science. DOI: 10.1126/science.2448875
Were you forwarded this newsletter?
LOVE IT.
If you liked what you read, consider signing up for your own subscription here: