Omic.ly Weekly 37
August 11, 2024
Hey There!
Thanks for spending part of your Sunday with Omic.ly!
I'll be on vacation from the 13th to the 25th. Unfortunately, edition 39 might be delayed until Tuesday the 27th, so don't be too worried if you don't get an email on the 25th.
Edition 38 will be loaded up before I head off on Tuesday!
This Week's Headlines
1) What came first, DNA methylation or the variant?
2) Everything you ever wanted to know about cluster based and single-molecule sequencing
3) Contrary to popular belief, there are 21 amino acids and the 21st was discovered in 1976
Here's what you missed in this week's Premium Edition:
HOT TAKE: Short-read sequencing on Nanopores ... just because you can doesn't mean you should, yet.
Or if you already have a premium subscription:
DNA sequence variants can change more than just the properties of the proteins that genes code for, they can also change what proteins get expressed!
This isn’t a new idea, we’ve known for a long time that DNA sequences in non-coding regions play an important role in regulating gene expression.
This is because these sequences can bind to transcription factors involved in gene expression, and many of these factors recognize specific sequences of DNA to perform these gene expression activities.
But we’ve come to appreciate that modifications to DNA sequences like DNA methylation are important for gene expression and highly methylated regions of DNA are usually turned off while unmethylated regions are turned on.
This is one of the basic concepts behind epigenetics, or the study of how non-sequence modifications affect gene expression.
What turns these regions on and off is a complex dance between proteins that methylate DNA, transcription factors that keep DNA ‘open’ by binding it, and the DNA sequence itself providing the recognition sites for these things to happen.
So, it goes without saying that the DNA sequence is an important component of this gene expression ballet, but we’ve struggled to fully understand how differences in methylation control gene expression.
In recent years we’ve discovered that our genome is 3 dimensional, and sequences very far away (in sequence distance) from genes can impact their expression because the genome can fold up on itself and bring these regions (and all the transcription activating factors they’ve bound) very close together.
But we’ve had a very hard time studying how DNA methylation in specific alleles (you have two copies of each gene) operate at a distance.
Fortunately, new long-read sequencing technologies are starting to change that because they can obtain sequence information across long-distances but they can also detect the methylation status of each allele independently.
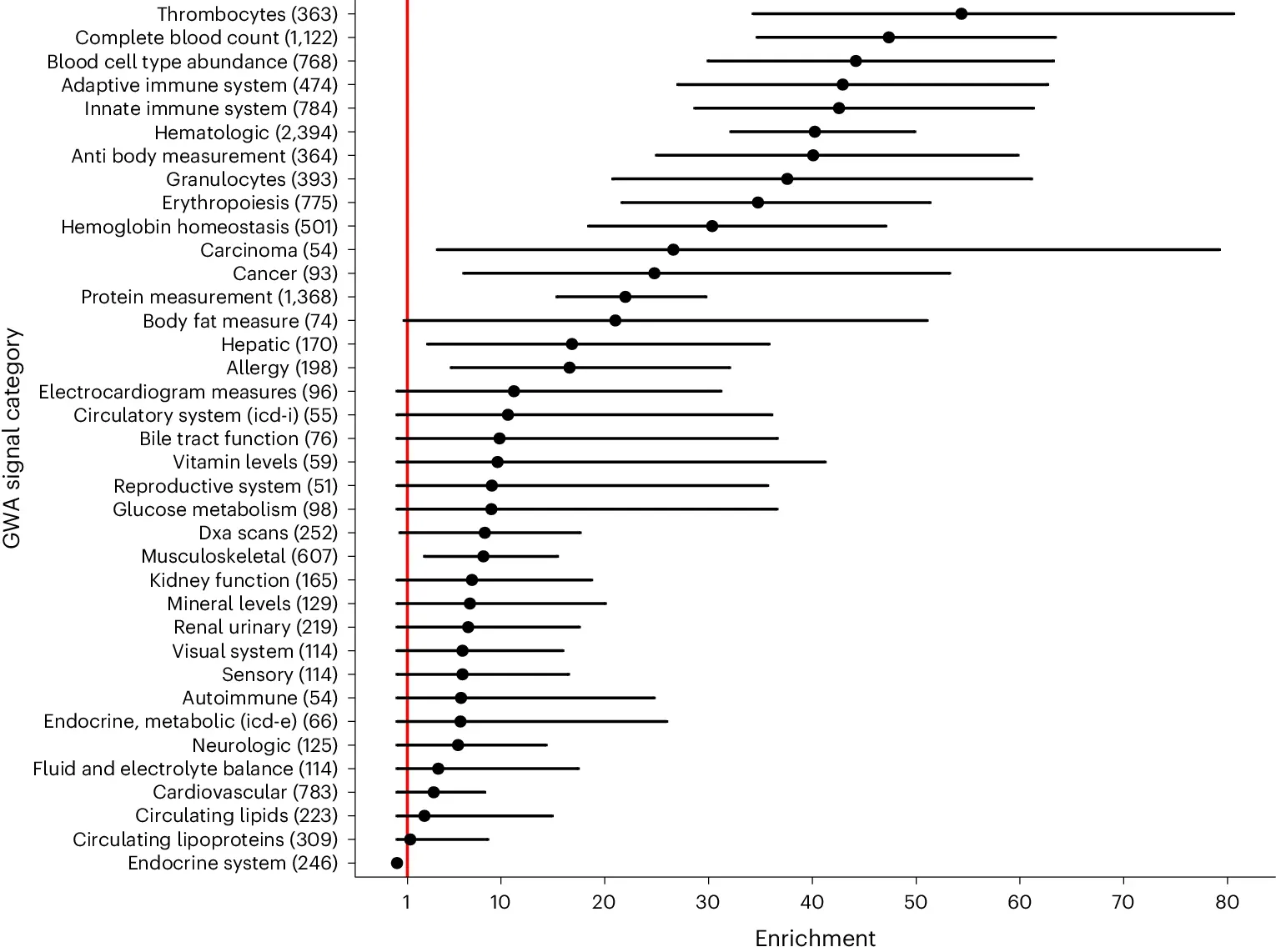
The authors of today’s paper used methylation-aware Oxford Nanopore sequencing to identify what they call allele-specific methylation quantitative trait loci (ASM-QTLs) in a cohort of ~7,000 human genomes.
If you’re not familiar with quantitative trait loci, they’re just regions of a genome associated with a phenotype or a trait.
They found that sequence variants were correlated with DNA methylation and RNA expression and that ASM-QTLs were enriched for a lot of traits (See figure above).
This is an exciting result because it confirms previous hypotheses that sequence variants that change DNA methylation at a distance can affect gene expression.
It also helps to explain how non-coding variants can modulate gene expression to help account for the wide range of phenotypic variability we see in health and disease.
###
Stefansson OA, et al. 2024. The correlation between CpG methylation and gene expression is driven by sequence variants. Nature. DOI: 10.1038/s41588-024-01851-2
Clusters vs single molecules in DNA sequencing: Here’s the short and the long of it.

There are 6 short-read companies and only 2 long-read companies.
Why is that?
Well, short-reads are cheaper so the market is bigger, and long-reads are really hard.
Short-read sequencing uses some version of random clustering, bead emulsion clustering, or whatever other slightly different iteration exists for patent purposes, but suffice it to say, cluster based sequencing is really only good out to about 400 base pairs.
This is due to phasing!
If you’ve done short-read sequencing, you know that phasing is an important metric, but you might not know why.
Basically, what happens when you short-read sequence is you start a little race at the beginning of each cycle.
And you do this with an amplified 'cluster' of many sequences that are clones from the same fragment so that you can efficiently detect the signal at each base!
The only problem is this race isn’t with people, it’s with cats. And cats don’t really care about your finish line so some get way ahead (Pre-phasing), some fall behind (phasing) and some all cross at the same time.
Luckily, the sequencing cats are nice and only a fraction of a percent of them misbehave every cycle – but if a fraction of a percent of them are lost every cycle, that limits how many cycles can be done, and you guessed it, that’s a couple hundred bases.
Very smart people have created algorithms to correct for this and have been able to push read lengths out to about 400 base pairs.
That’s why long-read sequencers don't use clustering but instead use very clever ‘single molecule’ schemes to generate data!
PacBio does this by watching an immobilized polymerase as fluorescently labeled bases are added to a single molecule. They use a powerful microscope and confocal techniques to view only the light emitted at the polymerase.
Alternatively, Oxford Nanopore Technologies (ONT) uses nanopores, and they sense the changes in current created as bases pass through the pore. Nanopore sequencing actually works by predicting the sequence content of a set of ~6 bases, a Kmer, from a single molecule.
But there's always an exception to the rule and there's one short-read company that dared to sequence single molecules.
That's Helicos Biosciences and since you've probably never heard of them you can guess how well this worked out.
They had a short stint in the spotlight before being eclipsed by Illumina in 2010.
However, their technology was resurrected in 2016 by SeqLL.
They've enjoyed about as much success as Helicos and their technology uses total internal reflection fluorescence (TIRF) microscopy to amplify and image the fluorescence from single molecules. While this sounds cool, they are limited to sub 60 base pair read lengths and don't have a broad user base.
ONT has also made noise about being able to do short-reads, but they're a very spendy option when it comes to short-read applications!
You might have been told that there are 20 amino acids. That’s a lie.
There are actually about 500 that occur in nature.
But only 22 of them are found in proteins across all organisms.
Of those, 21 appear in humans!
And I know it's killing you to know the name of the 21st amino acid.
But you're going to have to meet Thressa “Terry” Stadtman first.
Stadtman studied two anaerobic bacteria, Clostridium sticklandii and Methanococcus vannielii.
She isolated them from mud that she collected on the shore of San Francisco Bay in 1940.
They served her dutifully throughout her career.
She was fascinated by these microbes because they seemed to be like little chemical magicians!
They were able to perform chemistry at room temperature that would be nearly impossible in a traditional lab.
And they did this chemical magic using enzymes!
One of these was glycine reductase which converts the amino acid glycine to acetyl-phosphate (AcP).
AcP can recharge ADP and turn it into ATP.
If you don’t speak cellular metabolism, this means AcP can be used to literally make energy!
Stadtman was trying to figure out how this worked, but noticed that every so often her bacteria weren’t able to reduce glycine.
She discovered that this occurred whenever one of the components of her enzyme system was missing: protein A.
Thinking this was likely due to a deficiency in the food she used to grow her bacteria, she tested whether the addition of certain nutrients was able to fix the problem.
She found that dumping in Selenium, a trace element, restored protein A production!
But, that posed another question:
Why?
Stadtman decided to answer this by growing her bacteria on radioactive Selenium (75-Se) so she could see where it was going.
She discovered that it actually ended up IN protein A!
She published this finding in 1974, referring to protein A as a ‘selenoprotein.’
But this created another question:
Where was Se hiding?
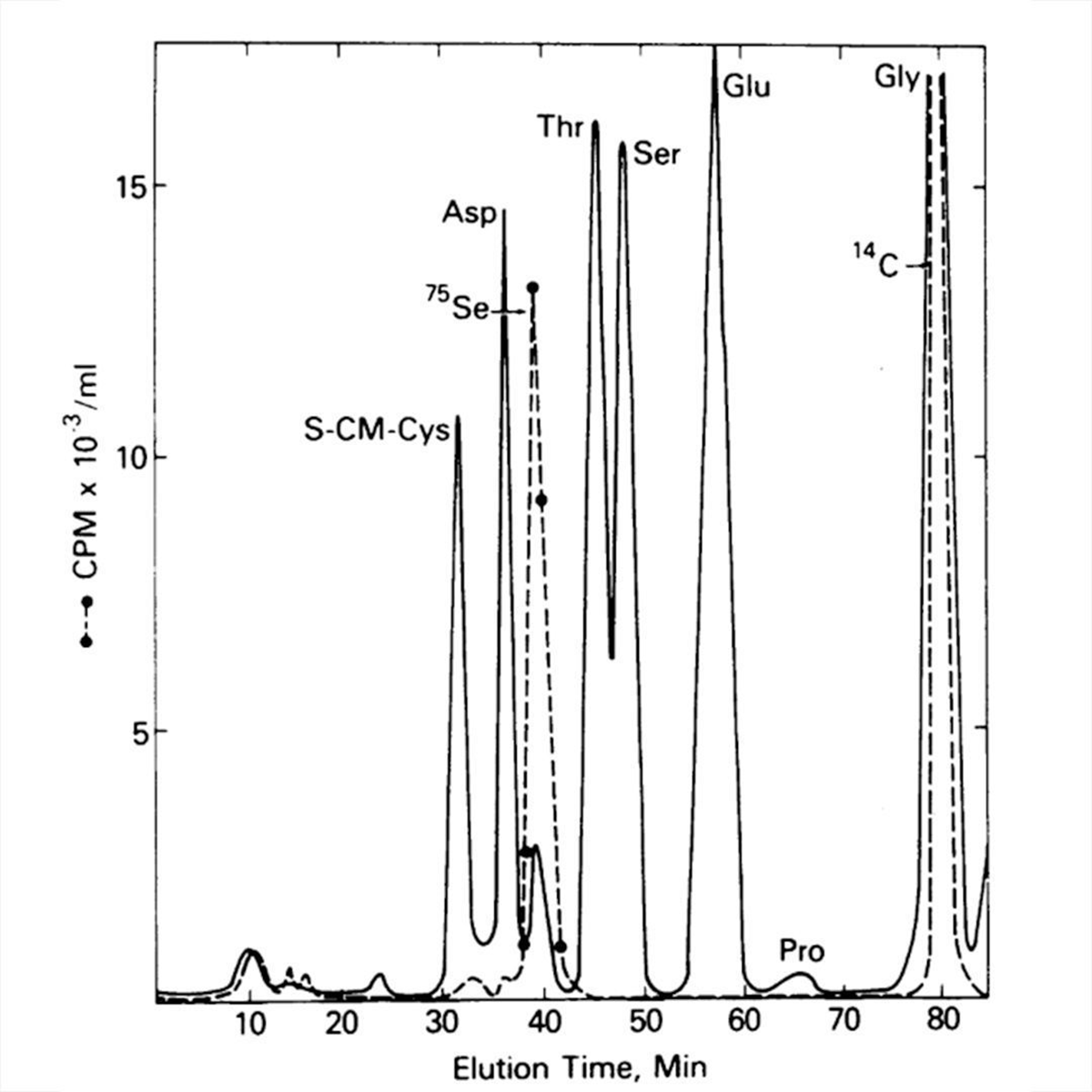
The figure above provides a pretty definitive answer.
Stadtman grew her bacteria on 75-Se again, isolated protein A, digested it down to its amino acid components, and threw that on a chemical analyzer!
In the plot, the solid line identifies the amino acid components of protein A and the dashed line tracks the radioactive signal.
Stadtman saw that the 75-Se peak coincided with a small unidentified amino acid peak after Aspartate (Asp).
The new peak was too close to Asp to be an Se derivative but she had a hunch that it was a new form of cysteine (S-CM-Cys) which appears first.
This was confirmed in a follow-up experiment!
Not only had Stadtman discovered where the Se went, she also discovered the 21st amino acid:
Selenocysteine!
It was later shown that this rare amino acid is coded for by the UGA stop codon and a special hairpin sequence at the end of a handful of RNAs.
###
Cone JE, et al. 1976. Chemical characterization of the selenoprotein component of clostridial glycine reductase. PNAS. DOI: 10.1073/pnas.73.8.2659
Were you forwarded this newsletter?
LOVE IT.
If you liked what you read, consider signing up for your own subscription here: