Omic.ly Weekly 35
July 28, 2024
Hey There!
Thanks for spending part of your Sunday with Omic.ly!
This Week's Headlines
1) How does the brain cause headaches if it can't feel pain?
2) Counting the things short-reads are good for
3) Green Fluorescent Protein was first discovered in Jellyfish in 1960
Here's what you missed in this week's Premium Edition:
HOT TAKE: Molecular Loop's dual barcode patent litigation should be giving everyone in the sequencing industry night terrors
Or if you already have a premium subscription:
Do you suffer from migraines? We might have some new clues about how they happen.
If you’re a migraine sufferer like me, you know the classic symptoms.
First, something just feels off, then you see blurry lightning bolts followed by a nasty headache that can last for hours (or days!)
Migraines aren’t fun and humans have suffered from them for basically our entire existence.
The earliest descriptions of them can be found in writings from ancient Egypt and the word migraine comes from Greek and means ‘pain in half of the head.’
We’ve come a long way in 3500 years, but still struggle to understand how migraines work.
60% of people suffer from prodrome (sensitivity to light, sound, etc), most people experience the aura phase (visual aberrations or odd sensory feelings), and almost everyone has the pain or headache phase.
Studies of the brains of migraine sufferers have shown that the brain undergoes Cortical Spreading Depression (CSD) at the onset of migraine symptoms.
This is just a fancy way to say that the brain experiences a period of hyperactivity followed by a wave of inhibition and it’s CSD that is most likely responsible for the weird feelings that are experienced during the aura phase.
But the pain phase of migraine is interesting because the brain itself doesn’t have pain receptors.
So, how does the brain tell the rest of the body there’s a problem if it can’t feel pain?
It seems to do this by signaling that pain through the peripheral nervous system and the nerves in our face, jaw and neck.
The authors of today’s paper show how that happens.
They discovered in mice that the central nervous system can signal to the PNS through cerebrospinal fluid.
They figured this out by staining CSF and watching through a microscope as the fluid flowed from the CNS to the trigeminal ganglion (the nerve bundle that transmits signals from the jaw and face).
This in itself is a huge development because signals from the CNS and PNS usually travel through the nerves, but since the brain doesn’t have pain receptors, that can’t happen!
So, evolution found a way.
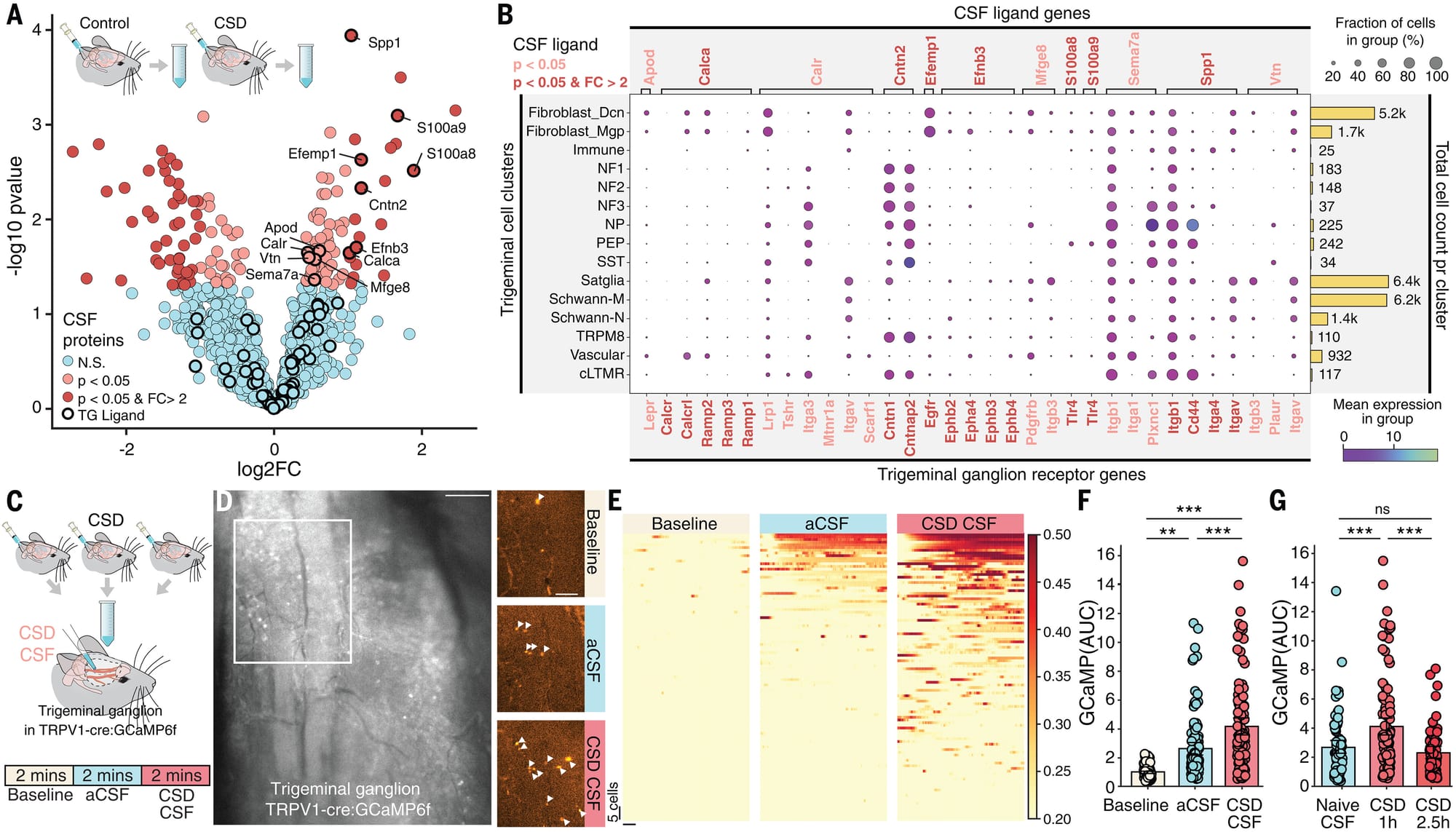
The authors then went on to sample the CSF to see what changed in it to signal this headache inducing pain response from the brain via the trigeminal ganglion.
What they found can be seen in the figure above: A) plot showing gene expression changes. B) Analysis showing how stuff in the CSF can signal to the cells of the trigeminal ganglion. C and D) Microscopic view of migraine CSF stimulating the ganglion more than an artificial control (aCSF). E, F, and G) Quantify stimulation and show that old CSF doesn’t have the same effect.
This work is important because it shows a novel mechanism for how the brain can trigger ‘headaches’ during migraine and offers new pathways to target for therapeutic development.
###
Rasmussen MK, et al. 2024. Trigeminal ganglion neurons are directly activated by influx of CSF solutes in a migraine model. Science Advances. DOI: 10.1126/science.adl0544
What's the point of short-reads if long-reads do basically everything in genomics better? There are five, FIVE, *thunderclap*, applications where short reads excel, ah, ah, ah!

One. Exon Target Capture:
Target capture sequencing is a technique that uses RNA or DNA probes to bind to and pull out specific sequences of interest. This is usually done for targeted gene sequencing applications all of the way up to full exome sequencing. Short-reads are ideal for this application because most coding regions of genes, or exons, are ~200bp. The standard 150x150 paired end technology is perfect for picking up these sequences without wasting sequencing dollars on introns or other untranslated regions of the genome.
Two. ctDNA:
Circulating tumor DNA sequencing methods are sometimes referred to as liquid biopsies. This method sequences the DNA that is shed by tumors into the bloodstream. These fragments of DNA are usually very short, in the 150-200bp size range and so are a perfect fit for short-read sequencing methods.
Three. cfDNA:
Circulating cell free DNA is similar to ctDNA except the distinction here is that cell free DNA doesn't originate from tumors, it originates from normal cells. This application is best known for its use in non-invasive prenatal testing where cell free DNA that's released by the placenta can be assayed to determine if a fetus has chromosomal abnormalities early in a pregnancy.
Four. FFPE:
Formalin Fixed Paraffin Embedded tissue is easily the worst sample type ever (gut microbiome/wastewater samples are a close second). The process of preserving biopsies back in olden times required placing a tissue sample into formaldehyde. To then look at these samples under a microscope, they were embedded in paraffin wax to make them easier to slice up and get onto a slide. While this is a reasonable process for microscopy, it makes getting usable DNA out of them a total pain in the ass. This means DNA ends up getting very fragmented which works in the favor of short-read sequencing methods! Maybe someday we'll be able to get physicians to update their methods to something more sequencing friendly, like flash freezing.
Five. Counting:
Counting applications are those where we're trying to figure how much of something is in a sample. Short-read sequencing is actually a really good high throughput multiplex counting method! That's especially true when using 25x25 - 75x75 chemistries which are the most cost efficient. Examples here are: counting sequence tags (like for proteomics), counting RNAs (expression profiling), bacteria/microbiome community profiling (genus level), differential methylation (liquid biopsy), chromosome counting (like in non-invasive prenatal testing), and epigenetic profiling.
So, short-reads will continue to have their place, but the go-to moving forward for clinical genomes and transcriptomes will be long-reads.
That's something you can count on! Ah, ah, ah!
In 1994, an obscure jellyfish protein was stuck into a worm. It has enlightened biology and medicine ever since.
But before we get to the genetically engineered worm, we first have to talk about the discovery of that jellyfish protein in 1960.
Aequorea aequorea, the jellyfish species in question, might seem like an odd place to start, but jellyfish are bioluminescent.
Osamu Shimomura was interested in studying the chemistry and physics of how these animals were able to produce their blue-green glow.
Up until that point, it was known that most bioluminescent creatures, like fireflies, do so with an enzymatic reaction between luciferin and luciferase.
But Shimomura discovered something different!
And in 1962 he isolated aequorin, a calcium binding protein that glows blue, along with another protein that had a bright green color under UV light.
Over the next decade, Shimomura dissected how aequorin and the green protein worked together to produce the characteristic glow of these jellyfish.
As it turns out, the blue calcium binding protein excites the green protein!
And in 1974 he published a paper describing what we now know of as green fluorescent protein (GFP).
Although Shimomura discovered the protein, he didn't see any further use for it and so focused his research elsewhere.
However, a few years later, this work was picked back up by Doug Prasher.
Unfortunately, getting a hold of enough protein to do anything useful with it was challenging because it had to be harvested from animals.
Prasher got the bright idea that he could use a new technique called cloning to make a bunch of this protein without having to collect and dissect a ton (literally) of jellyfish.
He was the first to sequence the GFP gene, but he couldn't get the cloned protein to light up, and so he gave up after publishing a paper about cloning the gene.
Soon after, Prasher was contacted by two labs who saw his paper and were interested in using GFP as a molecular tracer for imaging.
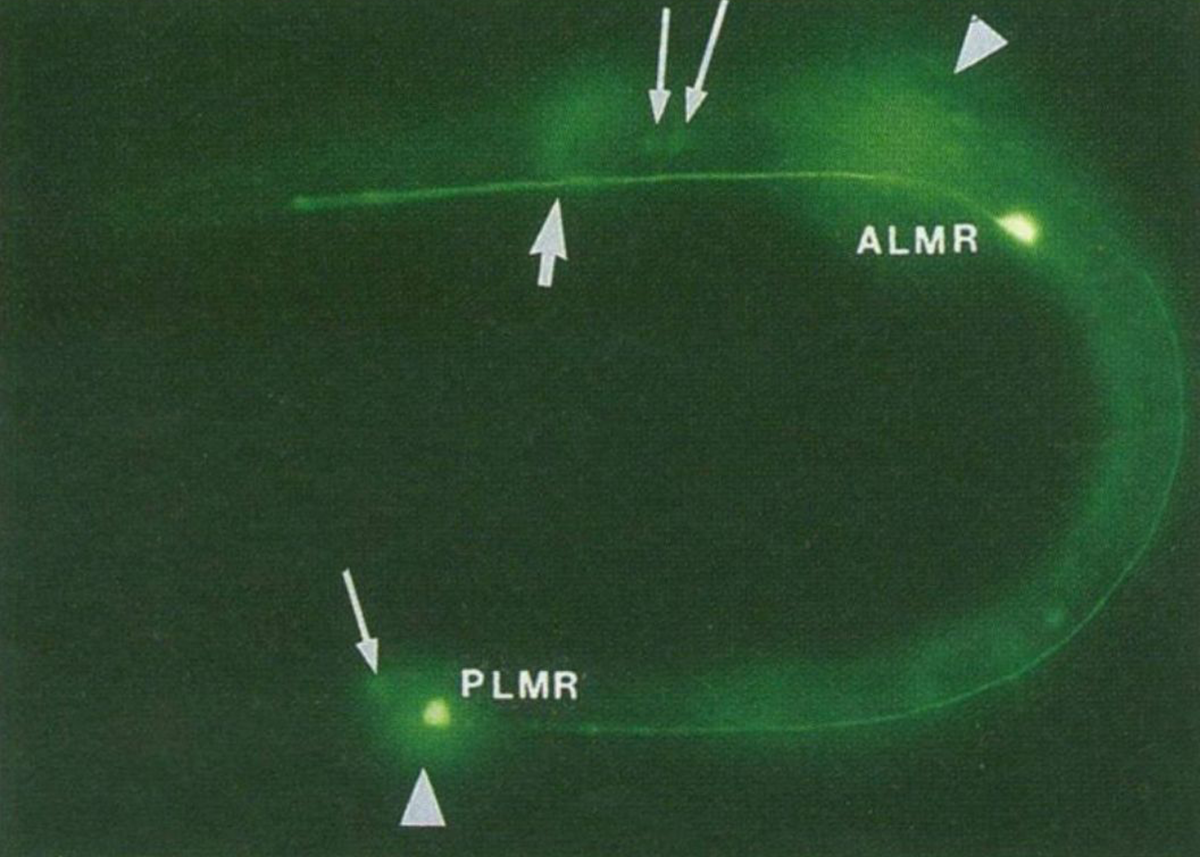
Martin Chalfie and his rotation student, Ghia Euskirchen, got the first shot and found that the original clone didn't work because it contained a couple of extra amino acids that prevented the protein from properly folding.
The figure above is the result of putting that now functioning sequence into C. elegans, a nematode worm.
It was expressed using an ALM/PLM neuron specific promoter (bright green dots).
But the story doesn't end there!
The second lab to get Prasher's GFP clone was Roger Tsien's.
Tsien was a biochemist and had visions for GFP beyond just green ones.
His lab created much of the fluorescent protein rainbow that we have available today.
Shimomura, Chalfie, and Tsien shared the Nobel Prize for this work in 2008.
And fluorescent proteins are now a ubiquitous molecular tool that color everything from cell cultures to aquarium fish!
###
Chalfie M, et al. 1994. Green fluorescent protein as a marker for gene expression. Science. DOI:10.1126/science.8303295
Were you forwarded this newsletter?
LOVE IT.
If you liked what you read, consider signing up for your own subscription here: