Omic.ly Weekly 34
July 21st, 2024
Hey There!
Thanks for spending part of your Sunday with Omic.ly!
This Week's Headlines
1) Reverse Translation doesn't exist in nature, but that's not stopping us from trying to do it!
2) Assay failures are common in the clinical lab, here are 5 of my favorites.
3) The Lac Operon: Everyone's heard of it, but do you know how it was discovered?
Here's what you missed in this week's Premium Edition:
HOT TAKE: There are a lot of companies working on protein sequencing, so who is in the lead?
Or if you're already a premium subscriber:
There are few things deserving of the superlative ‘holy grail,’ but Reverse Translation is one of them.
And that’s because it doesn’t exist in nature.
The Central Dogma states that DNA codes for RNA and RNA codes for protein.
But, proteins don’t serve as the template to code for other proteins, nor do they code nucleic acid.
We discovered in 1970 that some viruses can reverse translate RNA into DNA.
But the last bit about proteins has remained true since the Central Dogma was first postulated by Francis Crick in 1957.
That’s somewhat of a problem when it comes to being able to sequence proteins.
Because it means we don’t have any natural enzymes we can use to read back a protein sequence.
In DNA sequencing we have polymerases and reverse transcriptases that can spit out nucleic acid sequences, but we have nothing that can do that with proteins!
The current state of the art in protein sequencing uses ‘peptidases’ (enzymes that cut ‘peptide’ bonds) or Edman Degradation (a chemical reaction) that release amino acids at the end of proteins.
But in this scheme, detection is the hard part!
Some approaches fluorescently label cysteines and watch as the fluorescence signal decreases after every cycle of degradation.
This creates a cysteine sequence that can then be used to infer what protein was present.
Other approaches use antibody ‘recognizers’ to detect what amino acid is at the end of a protein before it’s degraded.
Neither of these approaches has scaled very well, they can’t detect every amino acid, and they struggle to recognize modified proteins.
With these issues in mind, the authors of today’s paper developed a method for performing reverse translation and sequencing of an immobilized peptide using DNA barcodes and antibodies.
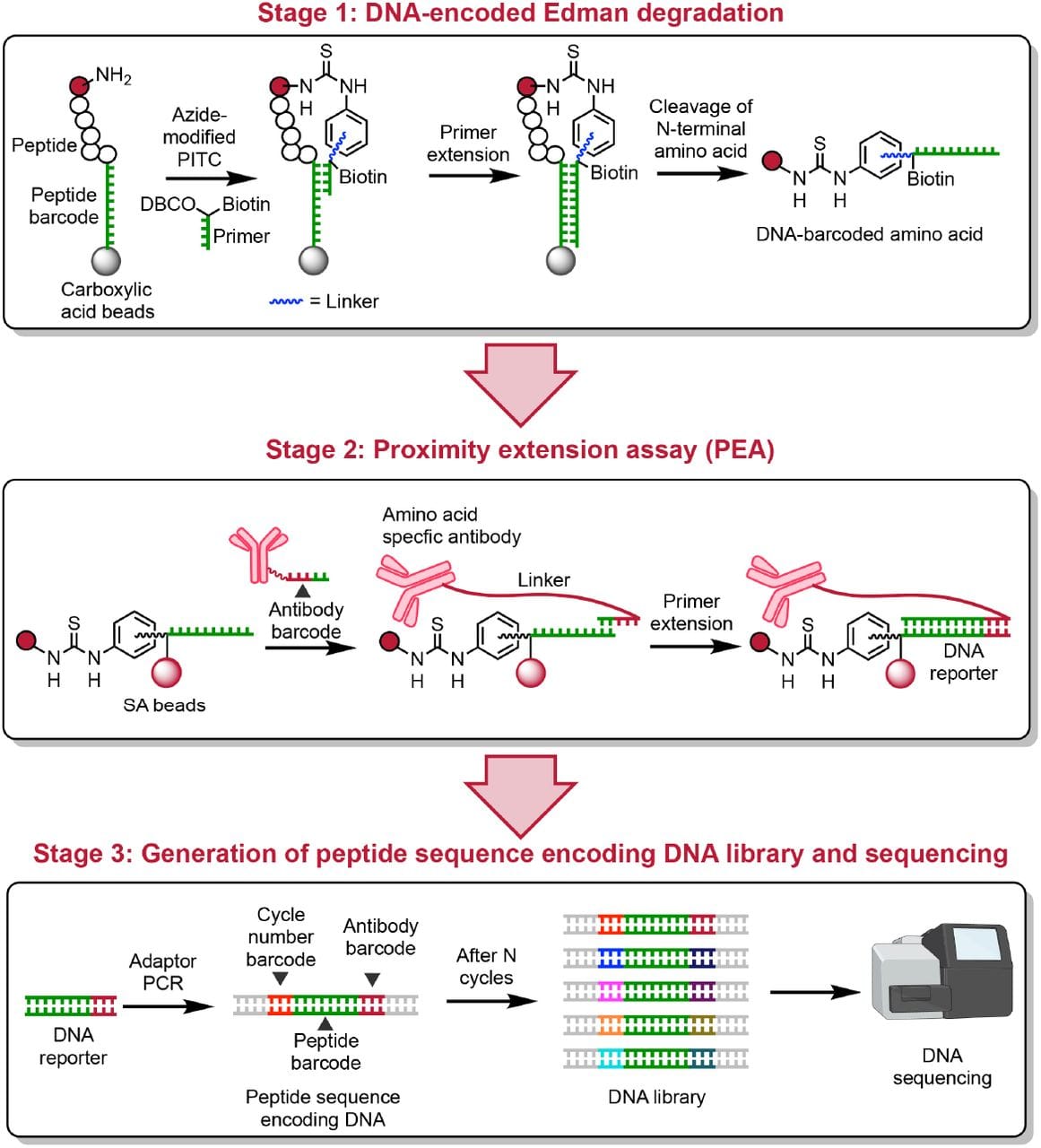
What they put together can be seen in the figure above.
Stage 1: A peptide is attached to a DNA barcode on its carboxy terminal end and exposed to a second oligo that binds the barcode sequence and covalently bonds to the amino terminal amino acid. A primer extension reaction is done to barcode the amino acid and then Edman Degradation is performed to release it from the complex. These barcoded amino acids are isolated every cycle.
Stage 2: The isolated barcoded amino acids are then exposed to oligo labeled antibodies that detect what amino acid is present. This adds an amino acid barcode and a cycle code to the DNA sequence.
Stage 3: The pieces of DNA from each ‘cycle’ of sequencing are combined, turned into sequencing libraries, and then sequenced on a DNA sequencer to determine the protein sequence.
The authors show that phosphorylation specific antibodies can be added at stage 2 to detect modified and unmodified amino acids.
While this process seems tricky to scale, it’s an interesting proof-of-concept that could be further developed into a real protein sequencer.
###
Zheng L, et al. 2024. Peptide sequencing via reverse translation of peptides into DNA. BioRxiv. DOI: 10.1101/2024.05.31.596913
Troubleshooting assay failures is a second full-time job in R&D. Here are my top 5 'favorite' root causes!

If you've worked in R&D, deployed a process into production, and had it running for any amount of time, you've probably been called in to troubleshoot a failure at some point.
Sometimes failures can be attributed to 'blameless' problems like instrument malfunctions or problems with a reagent lot.
Those are boring though, so I'm not going to talk about them and instead I am going to point fingers.
Some of my favorite root causes are as follows:
1) Changes to Temperatures or Timing - Processes are validated at specific temperatures, incubation durations and timing of steps. Even minor changes to any of these things can cause a process to fail. Too short and the process doesn't complete, too long and everything in your sample disappears, not the right temperature and nothing happens, incubated in a water-bath instead of a thermal cycler and your sample evaporates, try to setup 15 plates at a time and the first plates incubate way longer than the last plates. Timing and temperature are everything in molecular testing and shouldn't be changed without input and validation from the development team.
2) Reagent/Tip Waste Overflows - Empty the waste when it's full. Don't push it down because you think you have enough room to squeeze in another run, don't ignore the fill lines on the waste carboys, and definitely don't let any of these wastes overflow if they're flammable. This is especially important on robots where many deck components are electrified and can easily 'spark' a disaster if something like an ethanol waste container overflows!
3) Not Following Deck Setup Prompts - "The robot crashed and I don't know why." Did you follow the deck setup prompts to make sure you put everything in the right place? **Blinks** "No, I just click through all of those screens and set it up like I was trained." Le sigh. This is why we can't have nice things, people, and also why totally unnecessary scans and deck checks are added to automated processes just to be sure that when an important thing is listed in a prompt, that important thing actually gets done.
4) Repeat Pipettors - "It said pipette XX volume so I used a repeat pipette to do everything all at once." Please don't ever do this. Repeat pipettes have their place, for like bulk dispensing of wash buffers. They should not ever be used for activities where precision is required. For example, when setting up enzymatic reactions, pipetting concentrated buffers before dilution, or any other activity that requires very accurate volume dispensing.
5) Everything R&D Creates is Perfect, Except for When It's Not - 'Continuous improvement' is the mantra of the lab industry. We all try our hardest to do our best, but we can't know everything, and while it's fun to point fingers (even at ourselves!), these root cause analyses always lead to better processes!
'If gene expression determines the function of a cell, it must be important to control that process?' Yes, very! Let me tell you about the PaJaMo experiment.
Although we knew that DNA was the genetic material in the 1950s, there were still major questions about how DNA coded for protein and how that process, known as gene expression, was regulated.
While these were major questions at the time, an equally nagging question was whether enzymes (proteins) performed a specific function or if they could rearrange themselves to perform multiple functions.
There was a long standing hypothesis that enzymes were like little swiss army knives.
And they could be coaxed by environmental conditions to rearrange themselves to do different jobs depending on what was needed at the time.
A competing hypothesis argued that enzymes performed very specific functions and that it was actually the regulation of gene expression that determined how organisms responded to changes in their surroundings.
It was hypothesized that they did this by expressing different enzymes depending on what the current conditions required.
To settle this debate, Arthur Pardee, Francois Jacob, and Jacques Monod teamed up to study how bacteria are able to live and grow in the presence of multiple sugar sources.
It was known that E. coli could grow in the presence of both lactose and glucose and early work from Jacob showed that the 'lac' region of the bacterial genome seemed to control how they processed sugar.
This appeared to be the ideal system to suss out whether enzymes adapted to their conditions or if new enzymes were created to deal with whatever sugar was present.
Pardee, Jacob, and Monod (PaJaMo!) devised a series of experiments using bacteria that had different mutations (or combinations) in lac which contains 3 important regions:
z - makes beta-galactosidase, the enzyme that turns lactose into glucose and galactose
y - beta-galactosidase permease, the protein that brings lactose into the bacteria
i - inducer, a regulatory region that controls the expression of z and y
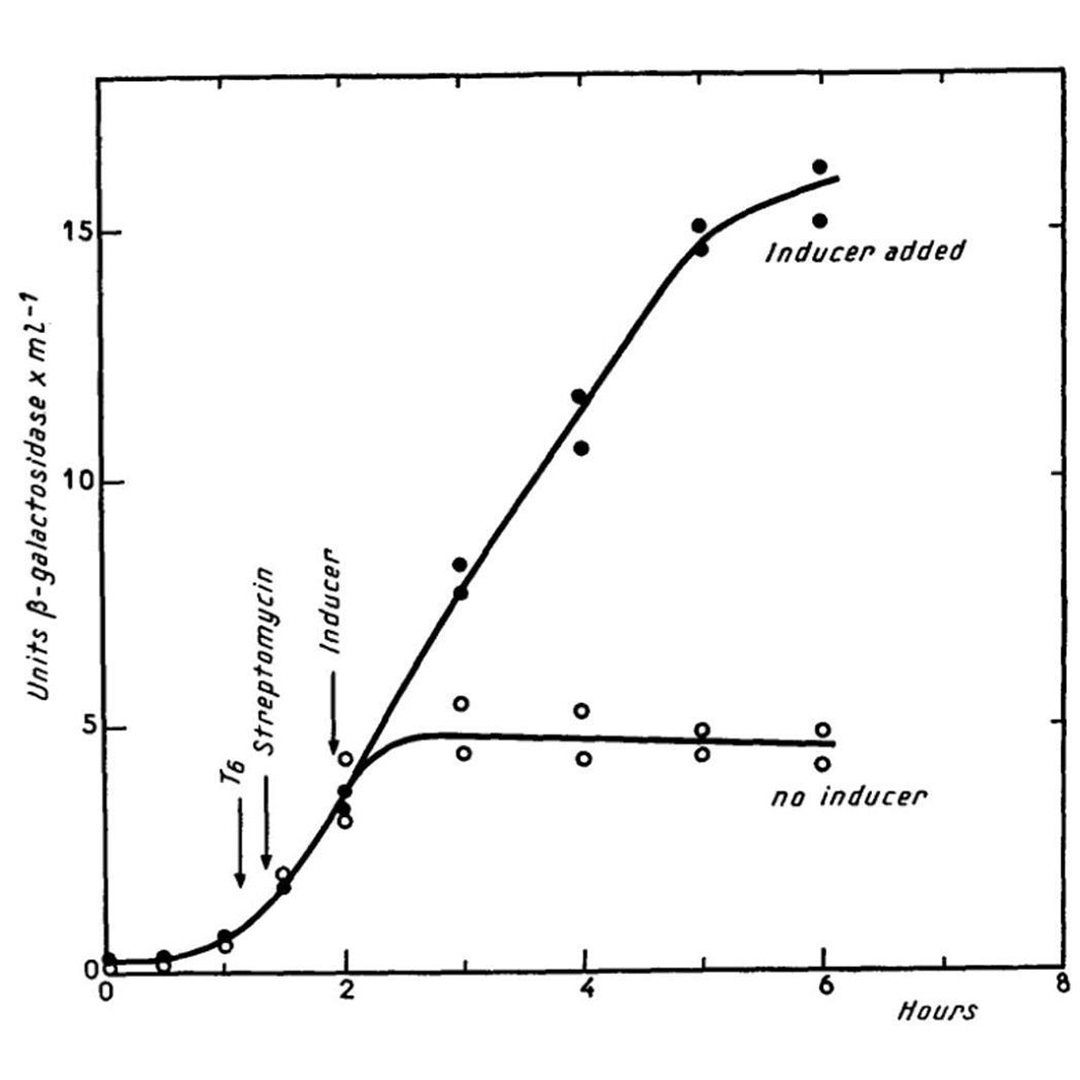
The figure above shows what happens in E. coli in the presence or absence of lactose (inducer).
When lactose is present (black dots) the bacteria make a lot of beta-galactosidase enzyme, when lactose isn't present (open dots) they don't make the enzyme.
Further research showed that the 'i' region is actually bound by a repressor protein that prevents the expression of z and y, and it's the binding of lactose to the repressor that releases this block!
While seemingly simple, the PaJaMo experiment unveiled what we now call the 'lac operon.'
It provided our first evidence that enzymes perform a specific function and that gene expression can be regulated to determine which enzymes are active at any given time!
###
Pardee AB et al. 1959. The genetic control and cytoplasmic expression of “Inducibility” in the synthesis of β-galactosidase by E. coli. JMB. DOI: 10.1016/S0022-2836(59)80045-0
Were you forwarded this newsletter?
LOVE IT.
If you liked what you read, consider signing up for your own subscription here: