Nanopores are getting on the rapid NICU/PICU sequencing bandwagon
Nanopores take on rapid neonatal sequencing
When it comes to diagnosis of rare, early on-set genetic disease, speed to diagnosis can make a huge difference in a child’s quality of life post-diagnosis.
This is why we’ve seen an explosion of stories (and studies) about rapid neonatal sequencing.
Much of this work was (and still is) championed by Dr. Stephen Kingsmore of Rady’s Children Hospital who debuted a 26hr genome in 2015 using a modified Illumina HiSeq 2500 to rapidly diagnose children in the Neonatal Intensive Care Unit (NICU).
But, one of the major limitations of short-read genome sequencing is that it misses things.
This is especially true with respect to mutations in pseudogenes, paralogs, variants that occur in repetitive sequences, and larger structural variants like inversions and translocations.
While short-read bioinformatic technologies have improved over the years, they’re still not as good at picking up all of the variant classes that we care about.
This means that kids who are not diagnosed with short-read whole genome sequencing still have to endure a diagnostic odyssey to get a final diagnosis.
This is usually done using follow up testing with things like microarray, methylation sequencing, or Multiplex ligation-dependent probe amplification (MLPA) to detect structural variants in problematic genes.
One way around needing to do all of this extra work in undiagnosed cases is to use long-read sequencing technologies such as those offered by Pacific Biosciences or Oxford Nanopore Technologies.
These sequencers look at multi-kilobase long fragments, and can also natively detect methylation patterns.
However, up until just a few years ago, these long-read technologies were prohibitively expensive and error prone.
Today, they’ve caught up with short-reads on both cost and quality.
And because they can access longer range information and detect methylation, they can diagnose cases that short-reads would miss!
So, why aren’t we using these superior technologies in the clinic?
That’s mostly because short-reads have about a decade long head start, and adoption of new technology in healthcare is excruciatingly slow.
The way we get around this is to generate clinical evidence to show that long-reads perform as well as or better than short-reads by comparing their performance in the clinic!
We’re just starting to see these studies come out now, and one was published last week.
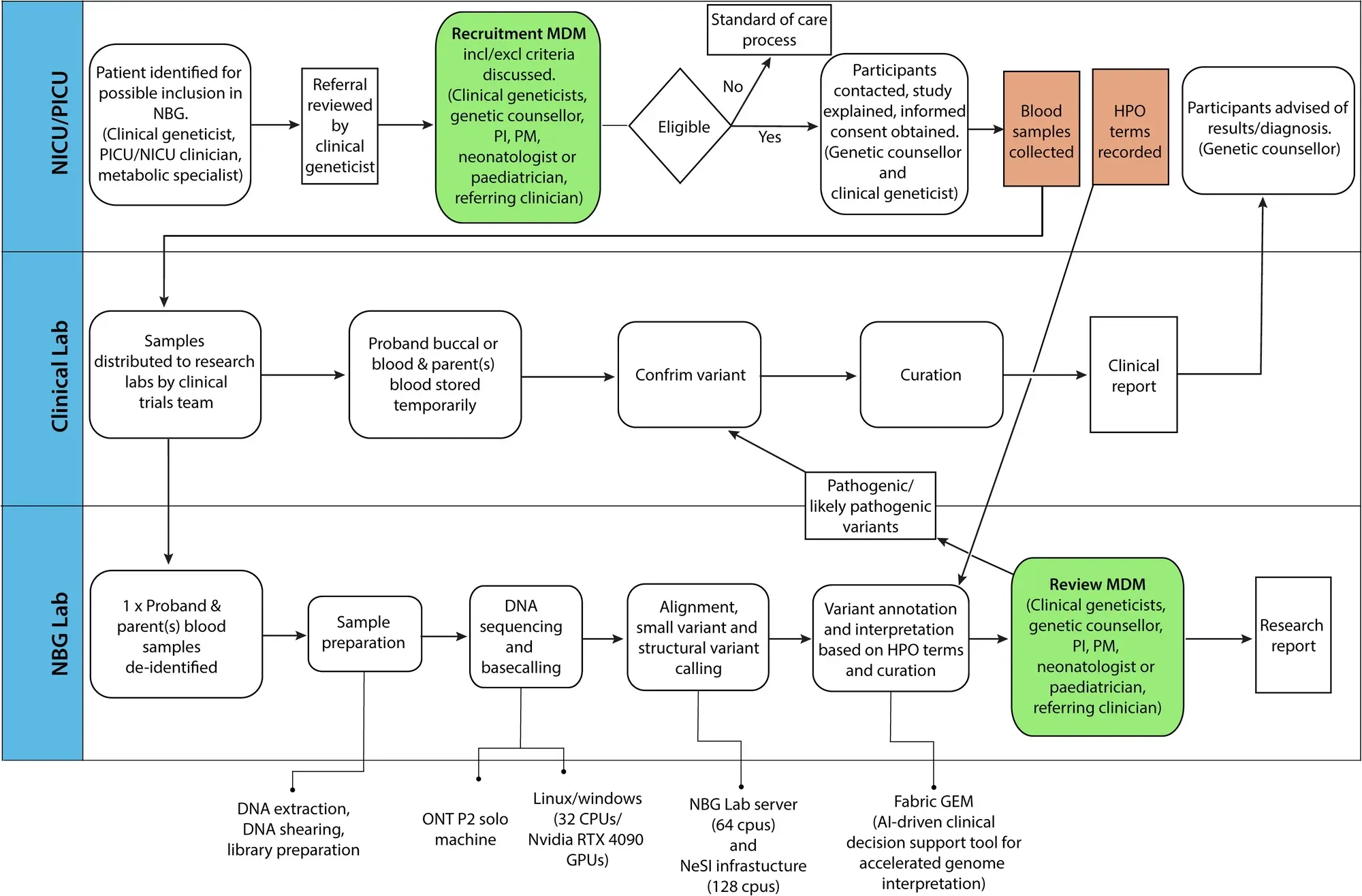
In this study, the clinicians used Oxford Nanopore sequencing on a P2 solo device to benchmark the performance of the sequencer against 6 well characterized genomes (HG002-7) and showed high levels of precision and recall for single-nucleotide variants (SNVs) (>.99) and small insertions and deletions (Indels) (>.8).
They also showed that the native methylation calls had high concordance with whole genome bisulphite sequencing on the same samples.
Finally, they developed a pipeline (which can be seen in the figure above) to perform rapid sequencing of neonatal patients in their NICU and showed a comparison of the first 10 results obtained using the rapid nanopore based method and more traditional Illumina short-read sequencing.
In the 10 cases presented, the rapid nanopore sequencing identified the same variants as the short-read method and diagnoses were obtained in 6 of the patients sequenced.
While this is a small study and not a full clinical validation of a nanopore based diagnostic pipeline, it’s a promising step in the right direction to help bring long-reads into clinical practice.