What came first, DNA methylation or the variant?
DNA sequence variants can change more than just the properties of the proteins that genes code for, they can also change what proteins get expressed!
This isn’t a new idea, we’ve known for a long time that DNA sequences in non-coding regions play an important role in regulating gene expression.
This is because these sequences can bind to transcription factors involved in gene expression, and many of these factors recognize specific sequences of DNA to perform these gene expression activities.
But we’ve come to appreciate that modifications to DNA sequences like DNA methylation are important for gene expression and highly methylated regions of DNA are usually turned off while unmethylated regions are turned on.
This is one of the basic concepts behind epigenetics, or the study of how non-sequence modifications affect gene expression.
What turns these regions on and off is a complex dance between proteins that methylate DNA, transcription factors that keep DNA ‘open’ by binding it, and the DNA sequence itself providing the recognition sites for these things to happen.
So, it goes without saying that the DNA sequence is an important component of this gene expression ballet, but we’ve struggled to fully understand how differences in methylation control gene expression.
In recent years we’ve discovered that our genome is 3 dimensional, and sequences very far away (in sequence distance) from genes can impact their expression because the genome can fold up on itself and bring these regions (and all the transcription activating factors they’ve bound) very close together.
But we’ve had a very hard time studying how DNA methylation in specific alleles (you have two copies of each gene) operate at a distance.
Fortunately, new long-read sequencing technologies are starting to change that because they can obtain sequence information across long-distances but they can also detect the methylation status of each allele independently.
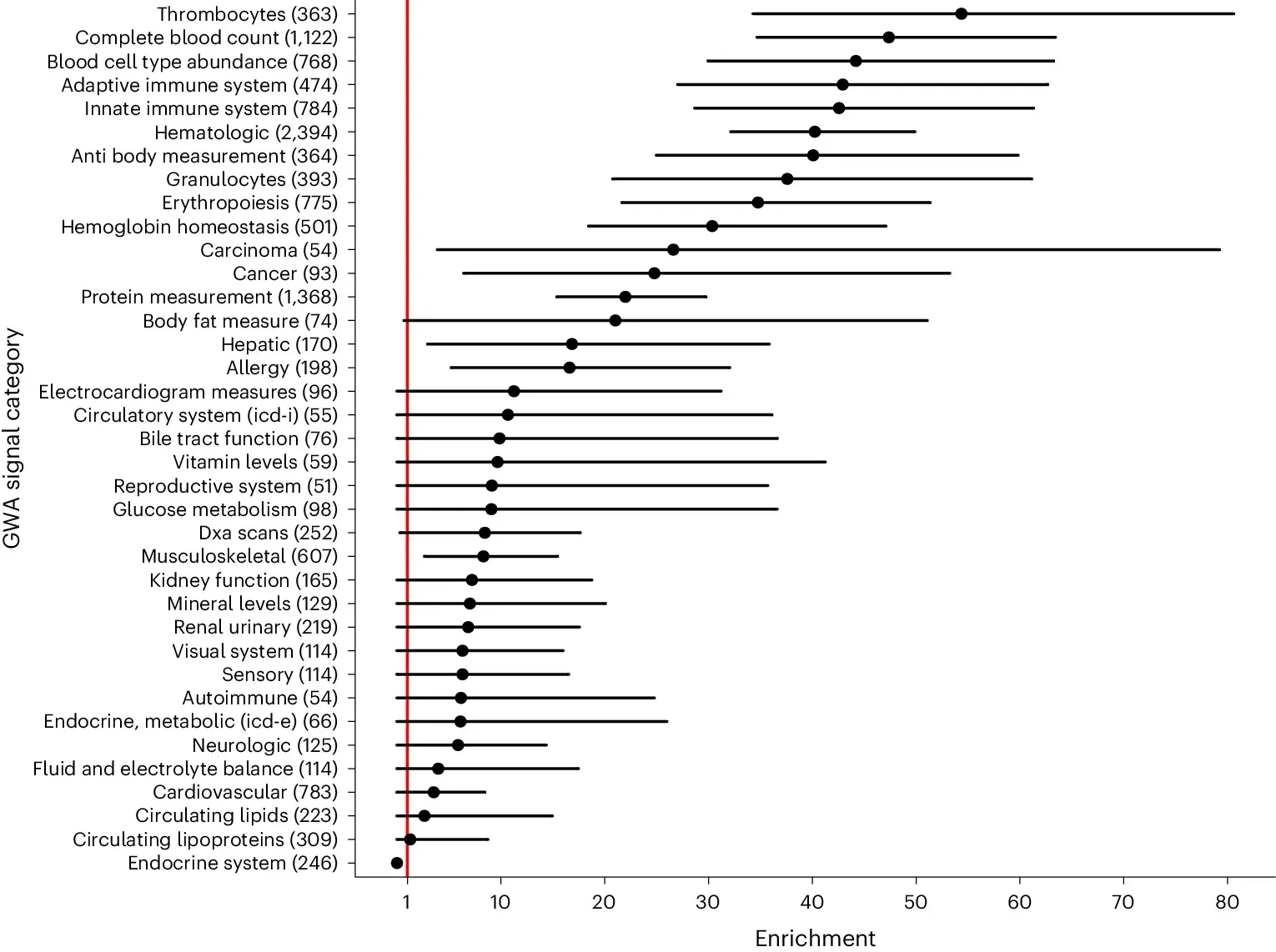
The authors of today’s paper used methylation-aware Oxford Nanopore sequencing to identify what they call allele-specific methylation quantitative trait loci (ASM-QTLs) in a cohort of ~7,000 human genomes.
If you’re not familiar with quantitative trait loci, they’re just regions of a genome associated with a phenotype or a trait.
They found that sequence variants were correlated with DNA methylation and RNA expression and that ASM-QTLs were enriched for a lot of traits (See figure above).
This is an exciting result because it confirms previous hypotheses that sequence variants that change DNA methylation at a distance can affect gene expression.
It also helps to explain how non-coding variants can modulate gene expression to help account for the wide range of phenotypic variability we see in health and disease.
 Brian Krueger
Brian Krueger