Complex genetic traits are getting taken to new heights
The genetics of height gets the whole genome treatment
Genetics can get complicated quickly, and that’s no secret when we’re talking about the genetics of height.
Sometimes we scientists try to over-simplify things in genetics.
We talk about mutations and variants like there’s a strict cause and effect relationship between them and some disease or phenotype.
This isn’t true for, like, most things - genetics is complicated as h e 🏒 🏒 more often than it’s not!
Even for things like Mendelian traits and diseases (where there is a clear gene-phenotype effect) there’s still variability in penetrance for most mutations.
This simply means that with respect to diseases, mutations can show variability in their severity, if they even result in disease at all.
But this ‘simple’ situation gets way more complex with *drum roll* complex diseases and traits!
Things like eye color and height are considered to be complex traits, meaning that they are controlled by the activities of multiple genes and/or environmental factors and do not typically follow Mendelian inheritance patterns.
Geneticists also refer to complex traits as being polygenic.
And for the longest time, most research done on complex traits used array genotyping to try to map all of the variants or regions in the genome that contributed to their presentation using genome wide association studies (GWAS).
In the example of eye color, we’re talking about what regions of the genome control color or shade, and with height we’re talking about…height or how tall someone is.
But complex traits get even more complex than just the mutations associated with the coding regions of multiple genes.
We’ve also learned in the years since our initial sequencing of the human genome that non-coding variants (things in enhancers, promoters or intergenic regions) can also play important roles in how genes are expressed!
Which means that for us to totally understand complex traits like height, we need to look at all variants in the genome that can contribute to a phenotypic effect.
Unfortunately, in our years of doing GWAS, we focused mainly on common variants in human populations that contributed to these traits which means we’ve missed a good chunk of the variation that contributes to the phenotypes that we see.
For height, it’s estimated we’re still missing 50% of that variation!
However, as the cost of sequencing has declined, that means we can stop just looking at common variants, and start using whole genome sequencing to look at rarer ones to help find that missing variation.
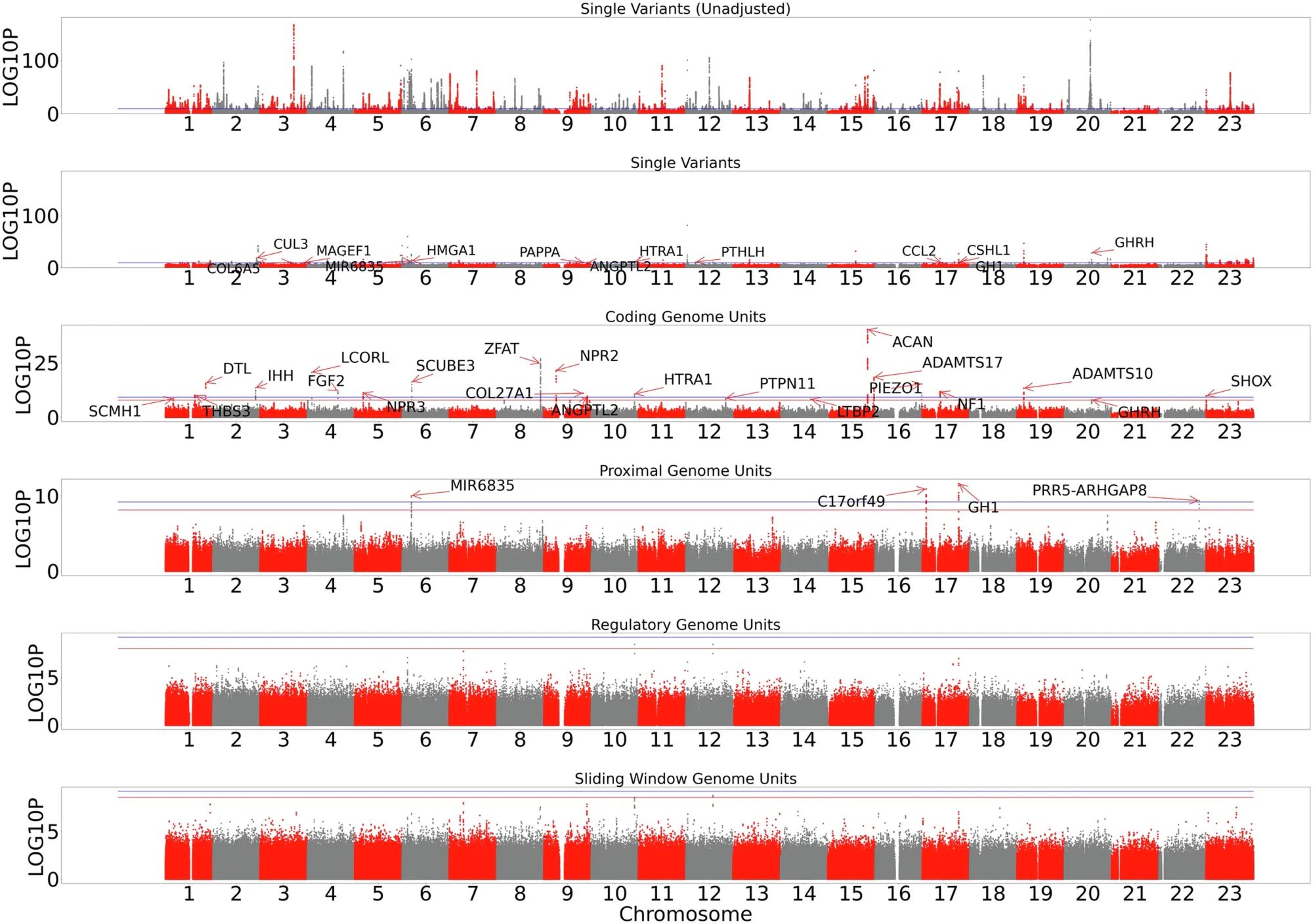
And that’s what can be seen in the figure above.
The researchers used whole genome data from the UK Biobank, All of Us, and TOPmed to suss out new genes and non-coding regions that control the variability we see in height.
The figure shows Manhattan plots for single variants, genes, proximal variants (within 5kb of a 5/3’ UTR), regulatory variants (more than 5kb from a gene), and whole genome sliding window (large regional contributions).
Locations were panel significant if they crossed the red line, or study wide significant if they crossed the blue line.
They found new single nucleotide variants or aggregate proximal variants associated with height in HMGA1, C17orf49, GH1, CSHL1, PRR5-ARGHGAP8 and MIR6835.
This study highlights the importance of considering the contributions of rare variants in complex traits for future genome wide association studies.