AI takes on virus evolution
AI to the rescue: using protein structural similarities to track virus evolution.
How do we know that organisms are related to each other?
Prior to the invention of DNA sequencing, a lot of that guess work was done by … guessing.
Basically, scientists in olden times looked at two organisms, saw what physical features matched between them, and then decided how related they were.
This, of course, is a gross oversimplification and actual measurements of the sizes of these structures and their appearance throughout the fossil record were used to place organisms into phylogenetic trees.
Charles Darwin famously created such associations using the characteristics of finch beak size on the Galapagos Islands to track the evolution of these birds from island to island.
But now that we’re in the age of genetics, we also include molecular genetic information during the process of creating these trees.
So, both physical characteristics and genetic characteristics are used to create the evolutionary trees that explain how something like a human shares a distant common ancestor with the great apes!
But how do we figure out the evolutionary history of things where their physical characteristics aren’t obvious, like in viruses?
Up until now it’s been based primarily on sequence similarity (and how they look under a microscope), but we have found these sorts of analyses to be challenging because viruses evolve (genetically) extremely quickly.
They have very short life cycles and are usually under extreme selection pressure ie immune systems don’t like viruses and try to kill them.
This means that viruses with little genetic tweaks that allow them to evade the immune system of their host can survive to live another day!
But this also means that their genomes can be a totally jumbled mess that makes it hard for us to figure out how they’re all related!
Wouldn’t it be cool if viruses had physical characteristics that we could use in concert with genetic data to help us with that detective work?
Well, viruses, like all other organisms, are basically just bags of nucleic acid and proteins.
As we’ve established, we can sequence DNA and RNA, so how can we use the proteins?
We could generate crystal structures of them from all of the viruses, but that would be an ungodly tedious task.
What if we could use protein folding AI to do that dirty work for us instead?
That’s the basic premise of this week’s paper where the authors went through an exhaustive characterization of the flaviviruses (includes all-stars like HepC, Zika, and Dengue) and found some exciting new relationships that would have been impossible to identify only using genetic sequences.
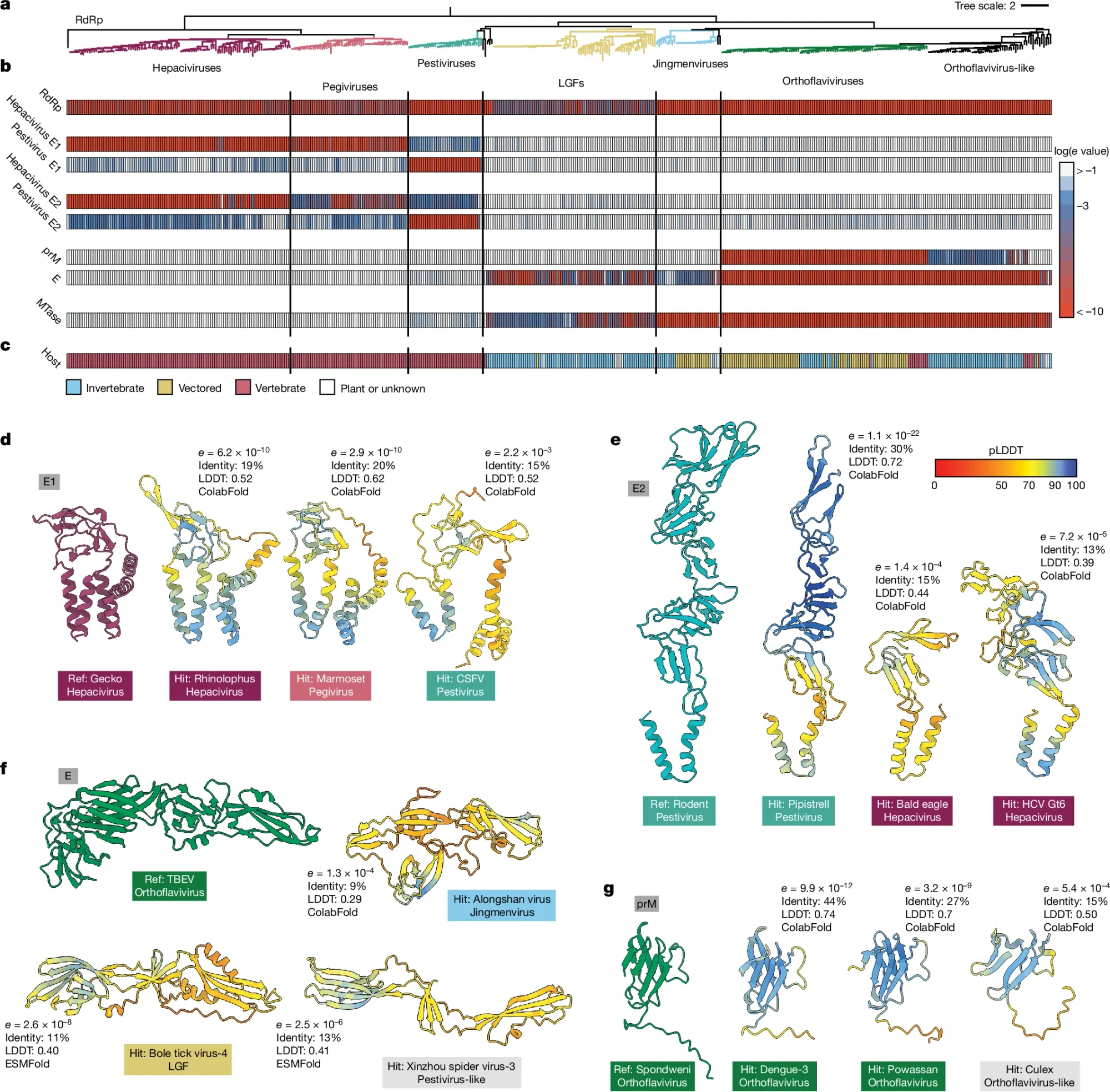
They did this by feeding sequences and protein structures into AlphaFold2/ColabFold and ESMFold to generate protein structures and then used FoldSeek to look for structural similarities among the proteins contained within the entire Flaviviridae proteome.
This can be seen in the figure above where in a) the researchers looked across all of the flaviviruses, b) chose a set of 8 proteins to compare for homology (Red - high homology, blue - low homology, white - no protein found), c) shows what host each virus infects, and d-g) show representative protein structure comparisons.
They then used these protein homologies to generate a new phylogenetic tree for the flaviviruses.
They also found that some flaviviruses stole a protein, Rnase T2, from bacteria!
While these results are scientifically interesting, they also have implications in the clinic.
Because, now we can use the knowledge that genetically distinct viruses might actually share similar protein structures and therefore might be susceptible to similar therapies!
But, this paper also highlights the importance of remembering that DNA and amino acids are like LEGOS, you can combine them in lots of different ways to make functionally similar things.
And that applies to all organisms, not just viruses!