A foundation model for transcription in all known cell types
A new foundation model has been trained that predicts transcriptional activity in all known cell types!
“But, does it make those predictions accurately?”
Shhhh, we’ll get to that, don’t spoil the fun!
First we need to define “transcriptional activity” which in itself is a crazy complicated topic.
But for brevity, transcription includes all of the things that operate together in the process of converting the genetic code into an RNA message.
This includes things like the binding of transcription factors to each other and to sequence motifs in DNA, the opening of chromatin to allow those things to bind, and the recruitment of an RNA polymerase to do the transcribing.
And it’s the transcription of DNA into an RNA message and its subsequent translation to protein that drives most of the activity in our cells.
Transcription is also one of the major factors that helps to determine the function of a cell and its role within a tissue.
To date, we’ve identified over 200 different cell types in our body and all of these interact to create the tissues and organs that make us who we are!
But how our DNA code can be manipulated to create all of these different cell types is still largely unknown.
We’ve made some progress figuring this out though, and new single-cell techniques combined with high throughput sequencing are providing us with more data than we’ve ever had on this topic.
And thanks to research consortiums like the Genotype-Tissue Expression project (GTEx), this data is all publicly available for researchers to dig into to develop new models of gene expression!
A number of gene expression models have been developed for specific cell types, but the researchers behind today’s paper decided to go the foundation model route (which attempt to make generalized predictions based on their training data).
What they produced was a model based on “chromatin accessibility data across 213 human fetal and adult cell types [that] accurately predicts gene expression in both seen and unseen cell types.”
They named it General Expression Transformer (or GET for short) and showed that it does a reasonably good job of predicting cell type specific gene expression (r2=.88 vs mean expression level - .6, transcription start site accessibility - .22, and gene activity - .26),
But making gene expression predictions in unknown cell types isn’t GET's only trick!
The researchers showed that it can also predict regulatory activity, identify cis-regulatory elements (the DNA sequence kind), and predict interactions between transcription factors.
That’s a lot of information to get out of a model trained ONLY on chromatin accessibility data!
But, what’s the point of a foundation model if you can’t show it actually does something useful?
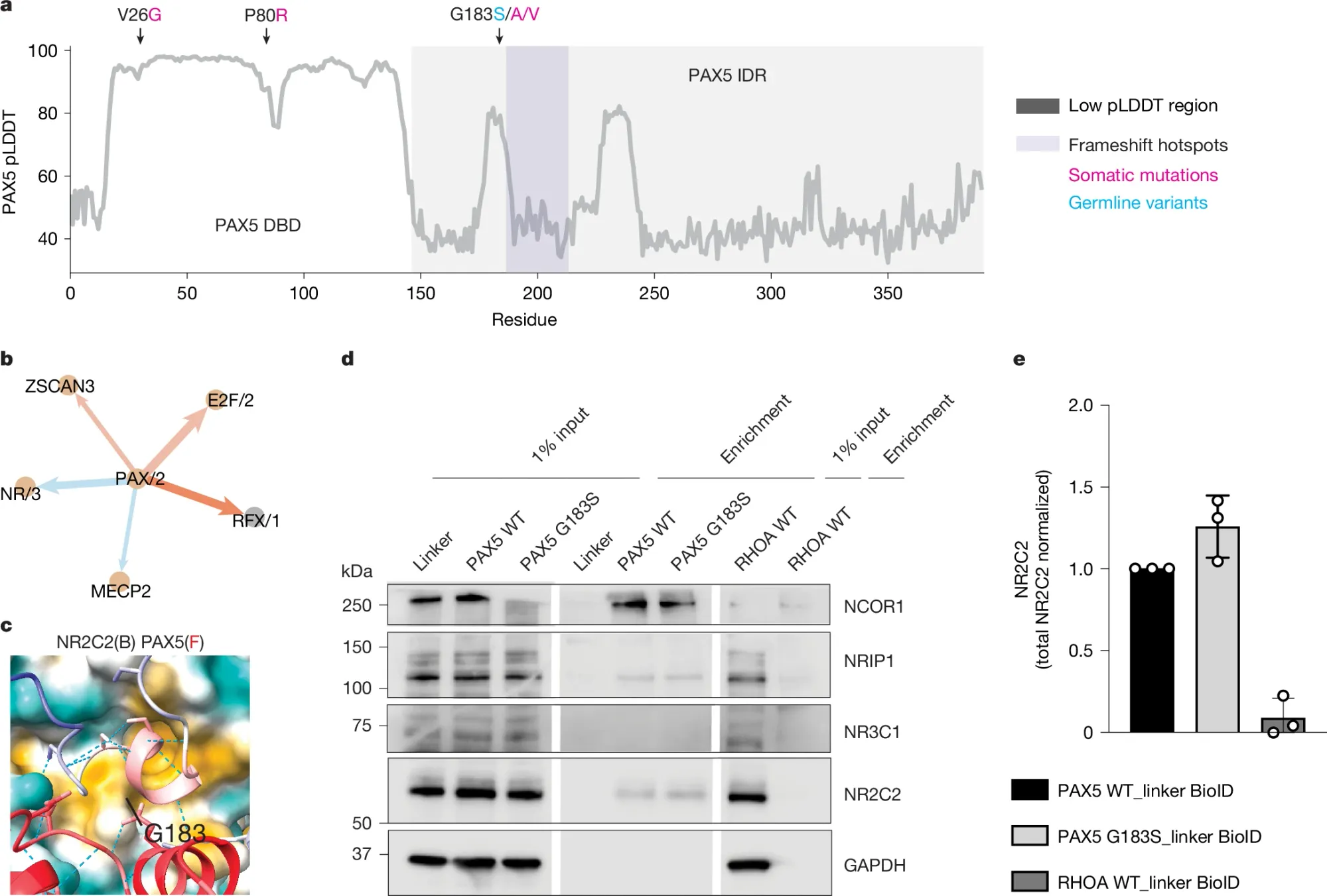
So, they put GET to work trying to figure out how a variant (G183S) in a transcription factor, PAX5, facilitates B cell precursor acute lymphoblastic leukaemia (B-ALL).
In a) they show that the G183S variant is in the intrinsically disordered region (IDR) of PAX5 b) GET predicted B-Cell sequence motif interactions includes PAX5 and NR/3, a common nuclear receptor domain c) AlphaFold predicted structure of the PAX5 IDR bound to an NR domain d) Binding experiment showing that PAX5 sticks (middle panel) to NCOR1, NRIP1, and NR2C2 e) The G183S PAX5 variant appears to make PAX5 bind better to NR2C2 (maybe…)
Despite the fact that all of these interactions were previously known, GET could be useful in prioritizing the follow-up of druggable pathways in diseased cells.
We can debate the utility of GET in its current iteration (I'm in the "meh" camp) but the authors suggest that training on more than just chromatin accessibility data (like perturb-Seq) will help to get GET better at predicting the transcriptional regulatory frameworks hiding within our DNA.